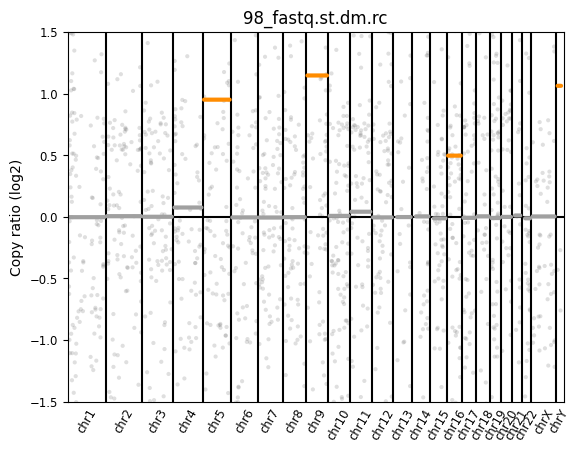
🔍 Copy Number Variation
- Germline copy number variants (CNVs)
- somatic copy number alterations (SCNAs)
🖇 PipeLine / Workflows
https://cnvkit.readthedocs.io/en/stable/pipeline.html
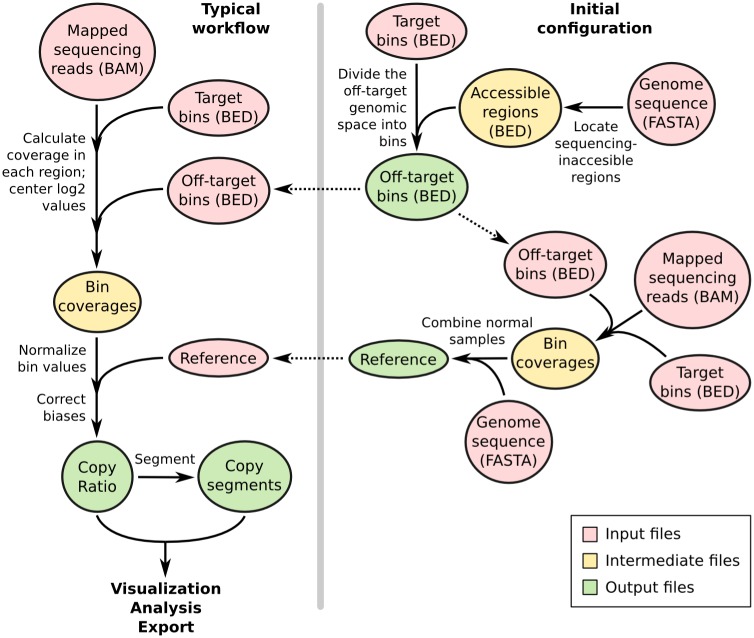
batch
=> automation of following processes
우선 input 으로 BAM 파일이 필요
< When matched normal sample, reference are available >
# From baits, and tumor / normal BAMs
cnvkit.py batch *Tumor.bam --normal *Normal.bam \
--targets my_baits.bed --annotate refFlat.txt \
--fasta hg19.fasta --access data/access-5kb-mappable.hg19.bed \
--output-reference my_reference.cnn --output-dir results/ \
--diagram --scatter
# Reusing a reference for additional samples
cnvkit.py batch *Tumor.bam -r Reference.cnn -d results/
# Reusing targets and antitargets to build a new reference, but no analysis
cnvkit.py batch -n *Normal.bam --output-reference new_reference.cnn \
-t my_targets.bed -a my_antitargets.bed \
-f hg19.fasta -g data/access-5kb-mappable.hg19.bed추가적인 옵션은 이 두개이며 결과를 그래프로 시각화 해준다. 만일 원하지 않으면 따로 써주지 않아도 된다.
- -diagram : diagram을 그려주는 옵션
- -scatter : scatter plot을 그려주는 옵션
=> equivalent to independent processes
cnvkit.py access hg19.fa -o access.hg19.bed
cnvkit.py autobin *.bam -t baits.bed -g access.hg19.bed [--annotate refFlat.txt --short-names]
# For each sample...
cnvkit.py coverage Sample.bam baits.target.bed -o Sample.targetcoverage.cnn
cnvkit.py coverage Sample.bam baits.antitarget.bed -o Sample.antitargetcoverage.cnn
# With all normal samples...
cnvkit.py reference *Normal.{,anti}targetcoverage.cnn --fasta hg19.fa -o my_reference.cnn
# For each tumor sample...
cnvkit.py fix Sample.targetcoverage.cnn Sample.antitargetcoverage.cnn my_reference.cnn -o Sample.cnr
cnvkit.py segment Sample.cnr -o Sample.cns
# Optionally, with --scatter and --diagram
cnvkit.py scatter Sample.cnr -s Sample.cns -o Sample-scatter.pdf
cnvkit.py diagram Sample.cnr -s Sample.cns -o Sample-diagram.pdf< When matched normal is not available >
cnvkit.py batch *Tumor.bam -n -t my_baits.bed -f hg19.fasta \
--split --access data/access-5kb-mappable.hg19.bed \
--output-reference my_flat_reference.cnn -d example2/- -t option; target, my_baits.bed 는 준비된 파일이어야하는지?
- --split --access ; 'calculate the sequence-accessible coordinates' , 이건 cnvkit 폴더안에 있다, 근데 나는 hg38 이용예정, 이걸 써도 될지?
- output-reference ; 생성된 FlatReference
- -d ; output directory
target
Prepare a BED file of baited regions for use with CNVkit.
cnvkit.py target my_baits.bed --annotate refFlat.txt --split -o my_targets.bed
BED file -> baited genomic regions ~ maybe unequal size, --split option would divide to the size close to specified by --average-size
- my baits.bed
- refFlat.txt ; if BED file doesn't have short, informative names for each bait interval
- -mytargets.bed
access
Calculate the sequence-accessible coordinates in chromosomes from the given reference genome, output as a BED file.
If your reference genome is the UCSC human genome hg19, a BED file of the sequencing-accessible regions is included in the CNVkit distribution as data/access-5kb-mappable.hg19.bed. If you’re not using hg19, consider building the “access” file yourself from your reference genome sequence (say, mm10.fasta) using the access command:
cnvkit.py access mm10.fasta -s 10000 -o access-10kb.mm10.bed
=> use this file in the next step to ensure off-target bins (“antitargets”) are allocated only in chromosomal regions that can be mapped.if there are ohter known unmappable, variable, poorly sequenced regions that should be excluded
cnvkit.py access hg19.fa -x excludes.bed -o access-excludes.hg19.bed
- hg38 과 같이 주어져있는 Referece file 이 있을때, 이 ref으로부터 interval list를 포함한 genomic coordinates 가 기록된 bed 파일이 access 명령어를 통해 만들어짐
~/GATK_best_practice/cnvkit.py access /BiO/Share/Tools/gatk-bundle/hg38/Homo_sapiens_assembly38.fasta -o access.hg38.bedantitarget
Given "target" BED , derive BED file off-target/"antitarget" regions
cnvkit.py antitarget my_targets.bed -g data/access-5kb-mappable.hg19.bed -o my_antitargets.bed
autobin
quicly estimate read counts/ depth in BAM => estimate resonable on/off target bin size
cnvkit.py autobin *.bam -t my_targets.bed -g access.hg19.bed cnvkit.py autobin *.bam -m amplicon -t my_targets.bed cnvkit.py autobin *.bam -m wgs -b 50000 -g access.hg19.bed --annotate refFlat.txtThe BAM index (.bai) is used to quickly determine the total number of reads present in a file, and random sampling of targeted regions (-t) is used to estimate average on-target read depth much faster than the coverage command.
coverage
calculate coverage in given regions from read depth
pileup ~ - ountnone optioncnvkit.py coverage Sample.bam targets.bed -o Sample.targetcoverage.cnn cnvkit.py coverage Sample.bam antitargets.bed -o Sample.antitargetcoverage.cnn
reference
compile CN reference from given files, given reference genome ( -f) calculate GC content, repeat-masked proportion
( A reference should be constructed specifically for each target capture panel, using a BED file listing the genomic coordinates of the baited regions. Ideally, the control or “normal” samples used to build the reference should match the type of sample (e.g. FFPE-extracted or fresh DNA) and library preparation protocol or kit used for the test (e.g. tumor) samples.)cnvkit.py reference *coverage.cnn -f ucsc.hg19.fa -o Reference.cnn
BUT 나는 paired/pooled normal 이 없다! => "Flat" reference of neutral copy number
cnvkit.py reference -o FlatReference.cnn -f ucsc.hg19.fa -t targets.bed -a antitargets.bed1) extract copy number information from one / small # of tumor sample
2) create 'dummy' reference to use as input to batch command
3) evaluate suitablity for analysis by repeating CNVkit analysisfix
combine uncorrected target & antitarget coverage tables (.cnn), correct for biases in regional coverage and GC content
cnvkit.py fix Sample.targetcoverage.cnn Sample.antitargetcoverage.cnn Reference.cnn -o Sample.cnr
how it works ;
A weight is assigned to each remaining bin depending on:
1. The size of the bin;
2. The deviation of the bin’s log2 value in the reference from 0;
3. The “spread” of the bin in the reference.segment
infer discrete copy number segments from give coverage table
cnvkit.py segment Sample.cnr -o Sample.cns
call
Given segmented log2 ratio estimates (.cns), derive each segment’s absolute integer copy number using either:
cnvkit.py call Sample.cns -o Sample.call.cns cnvkit.py call Sample.cns -y -m threshold -t=-1.1,-0.4,0.3,0.7 -o Sample.call.cns cnvkit.py call Sample.cns -y -m clonal --purity 0.65 -o Sample.call.cns cnvkit.py call Sample.cns -y -v Sample.vcf -m clonal --purity 0.7 -o Sample.call.cns
cnvkit.py call Sample.cns -y -v Sample.vcf -o Sample.call.cns
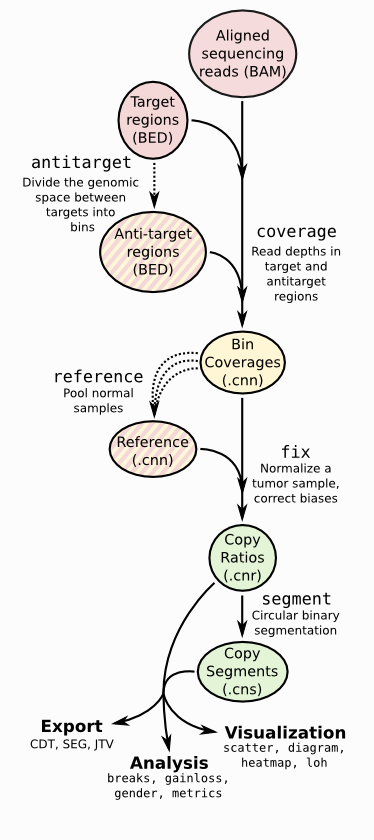
🗂 Review from Paper
Calculation of off-target intervals
- antitarget command ; accepts a list of targeted regions,
- Browser Extensible Data (BED) or GATK/Picard interval list format, and divides the off-target regions between each target into large bins
Estimation of copy number by read depth
Normalization of test samples to the reference
Segmentation and calling absolute copy number
Data summarization, reporting and visualization
