안녕하세요. dev_smapy입니다.
chemical을 다루는 library에 대해서 알아보려고 합니다.
바로 rdkit 인데요. 아실 분들은 이미 다들 아실거라고 생각합니다.
하지만 이제 막 이 분야에 관심을 갖기 시작한 분들은 '도대체 컴퓨터로 어떻게 화학을 한다는 거지?' 싶을 테니
이번 글에서는 rdkit을 어떻게 설치하는지와 간단한 사용에 대해서만 알아보도록 하겠습니다.
(파이썬을 사용하는 것을 가정하고 설명하겠습니다. 제가 파이썬 밖에 쓸 줄 몰라서..)
RDkit 설치하기
rdkit을 설치하는 방법은 간단합니다. conda를 이용하는 것입니다.
소스코드를 다운로드 받아서 설치할 수도 있습니다. 하지만 conda가 더 간단해요.
conda insteall -c conda-forge rdkit혹은
conda insteall -c rdkit rdkit으로 설치가 가능합니다.
RDkit 사용하기
설치를 했으니 사용을 해보겠습니다.
우선 파이썬을 들어가주세요.
pythonrdkit을 import 해줍니다.
import rdkit그런데 보통은 이런식으로 import 합니다.
from rdkit import Chem이렇게 기본적으로 설치와 사용을 할 수 있게 됐습니다.
여러 가지 chemistry file format
이제 rdkit을 사용해서 파일을 읽어보겠습니다.
읽기 전에, 일단 파일이 뭐가 있는지 알아야겠죠?
지금 자세히 다루지는 않겠지만, 자주 보는 format에 대해서 적어보겠습니다.
SMILES code
문자열 형태의 포맷입니다. 내부에 atom에 대한 정보와 bond정보를 갖고 있습니다.
3D에 대한 정보를 갖고 있지 않습니다.
확장자로 '.smi'를 사용하는 경우가 많고, 파일 안에 다수의 화합물을 포함하는 경우가 많습니다.
문자열로 구성하기 때문에 용량이 크지 않다는 장점이 있습니다.
예시) CCCS@c1ccc2[nH]/c(=N\C(=O)OC)[nH]c2c1는 다음과 같습니다.
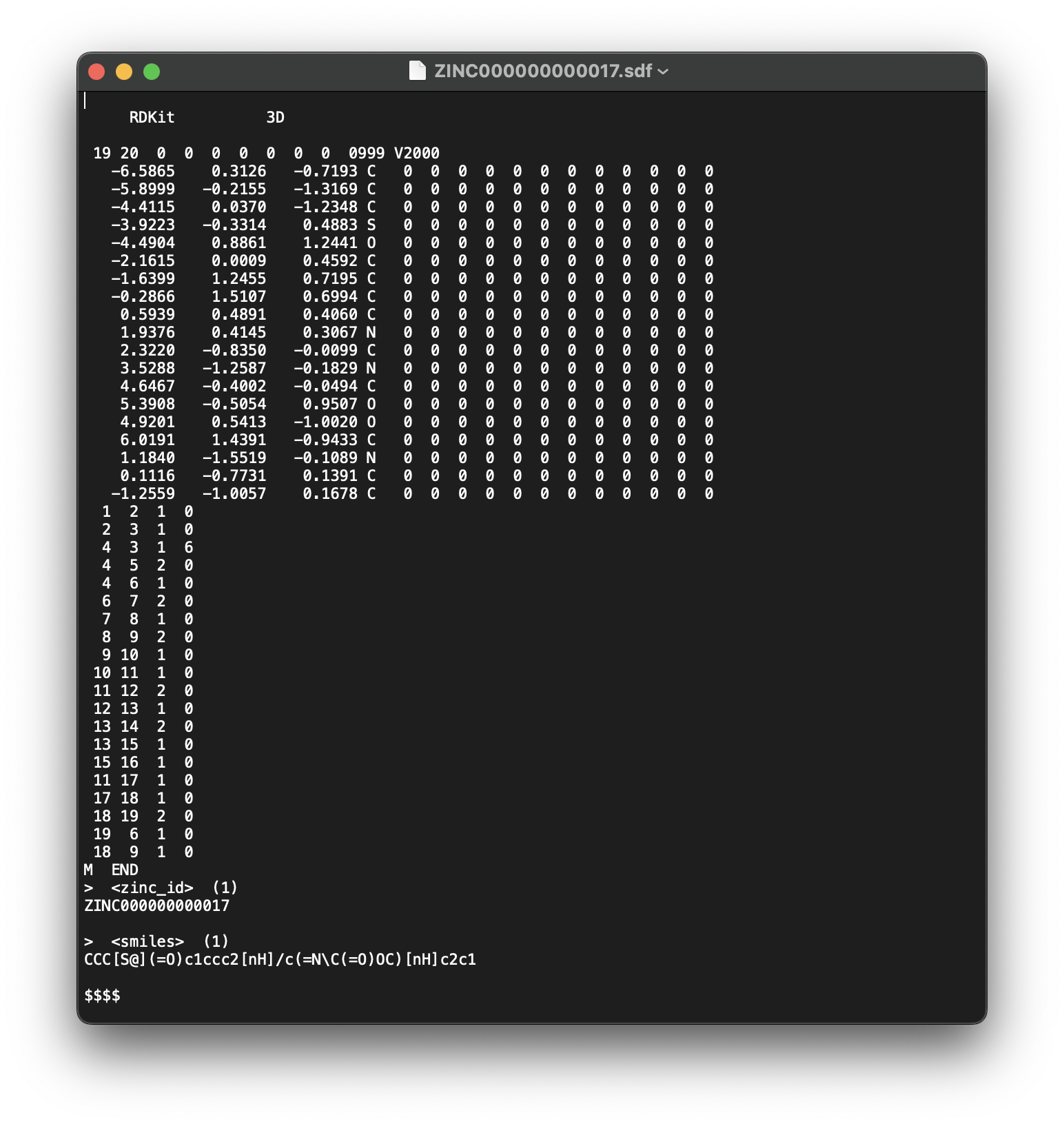
SDF file
3D 정보를 갖고 있는 포맷입니다. atom에 대한 정보, bond 정보와 같은 것을 포함하고 있습니다.
'$$$$'를 구분자로 여러 화합물을 연달아 포함할 수도 있습니다.
3D 좌표 정보를 포함하고 있어, 용량이 큽니다.
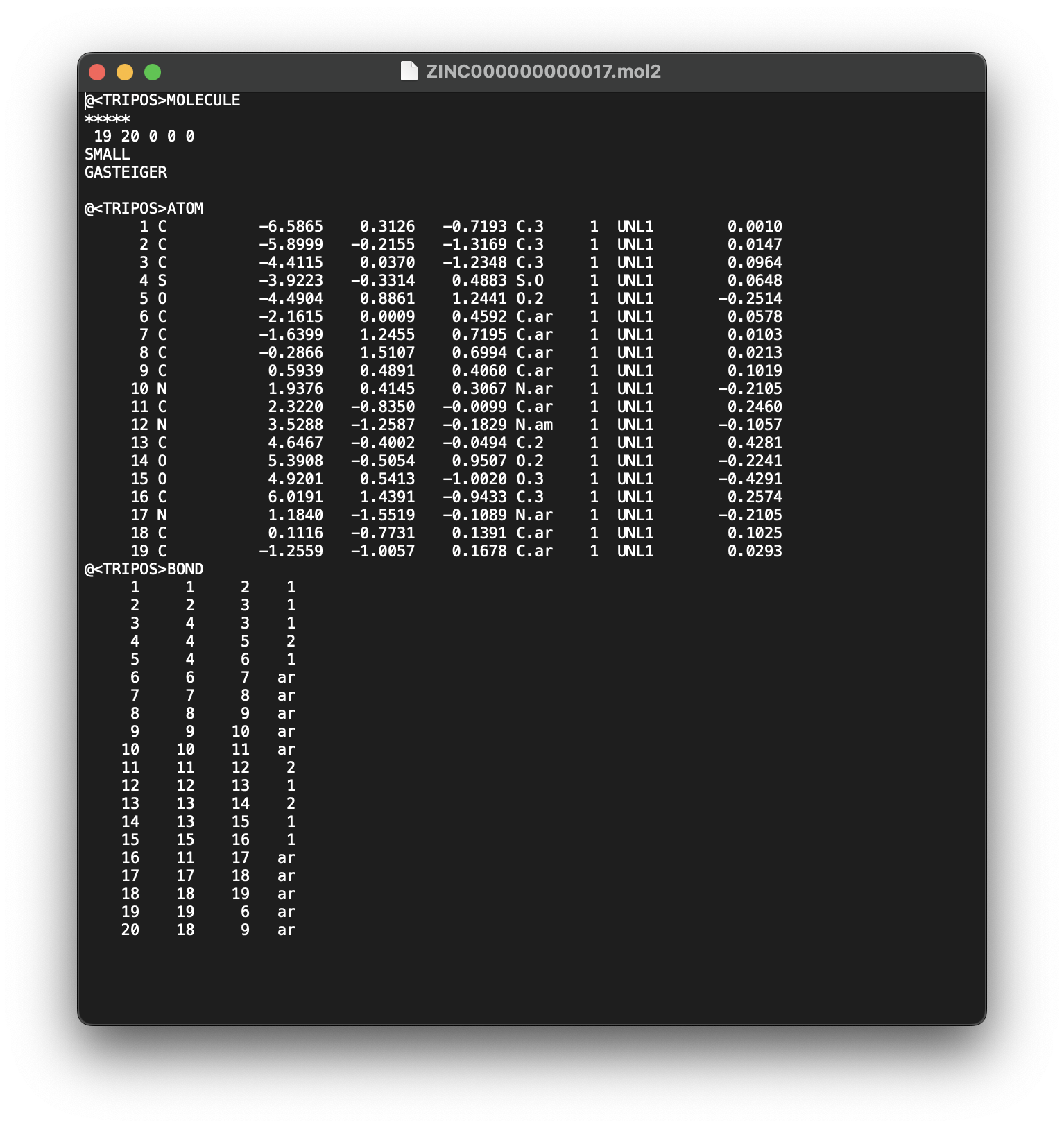
mol2 file
sdf file과 유사한 포맷입니다.
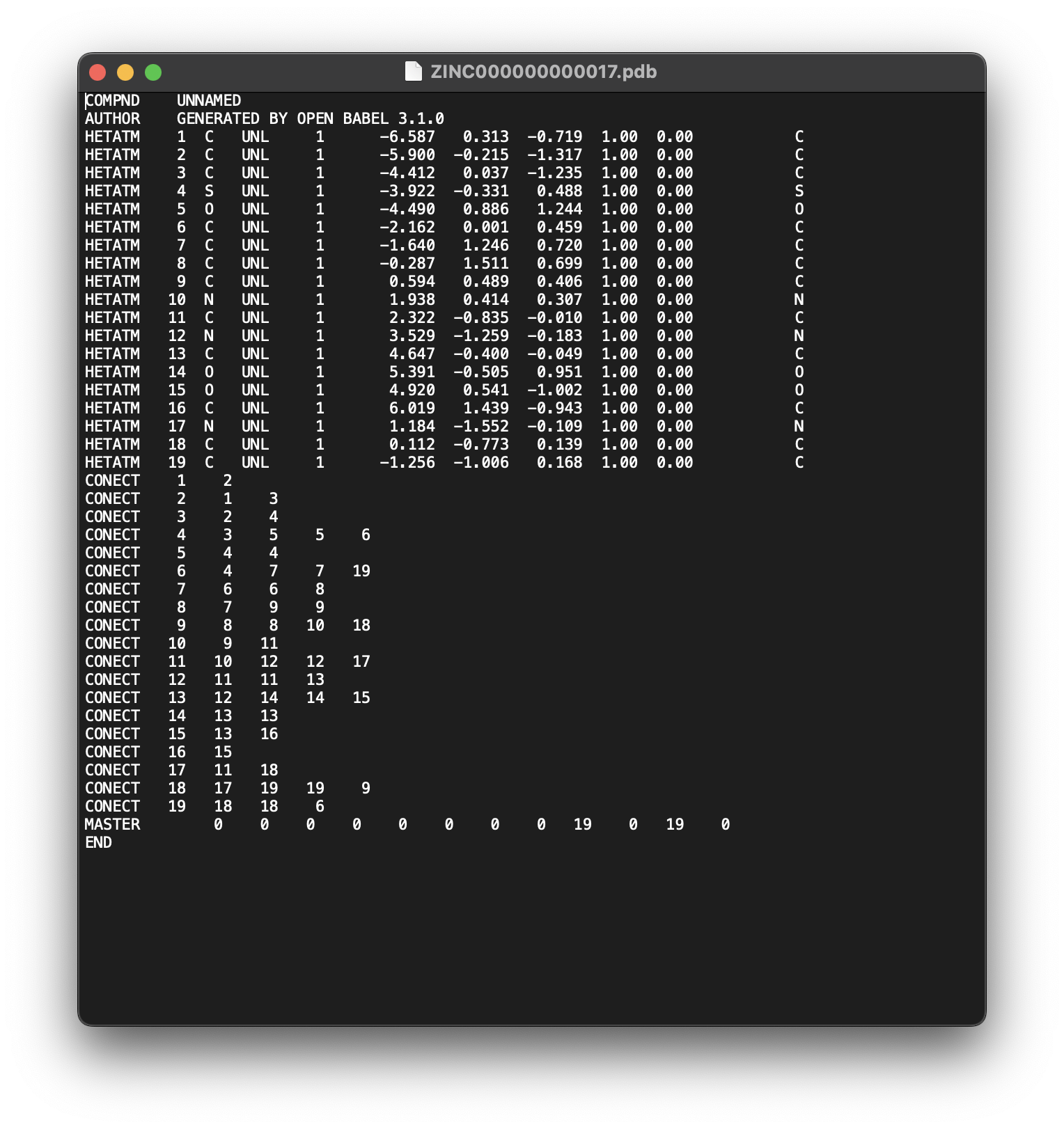
pdb file
sdf file, mol2 file과 유사합니다. protein data bank 포맷이고, column단위로 정보가 구별되어 적혀 있습니다.
TER를 구분자로 사용하고, 마지막에 END로 파일이 끝났다는 것을 표현합니다.
이미지는 모두 같은 화합물을 의미합니다.
RDkit을 사용해서 file 읽기
위에서 파일 포맷에 대해서 알아봤으니, 이제 rdkit을 통해서 읽어보도록 하겠습니다.
SMILES code
from rdkit import Chem
input_smiles = 'C1CCCCC1'
rdmol = Chem.MolFromSmiles(input_smiles)이렇게 문자열로 주어질 때, 읽을 수 있습니다.
파일 안에 다수의 SMILES code가 있을 때는 어떻게 할까요?
from rdkit import Chem
arr = []
with open('input.smi','r') as F:
for line in F.readlines():
tline = line.strip().split()
rdmol = Chem.MolFromSmiles(tline[0])
arr.append(rdmol)이렇게 읽어 주시면, arr안에 smiles를 rdkit으로 읽은 object들이 들어가게 됩니다.
SDF File
from rdkit import Chem
input_sdf = 'input.sdf'
rdmol = Chem.SDMolSupplier(input_sdf)이렇게 파일을 읽을 수 있도록, 내부에 메소드가 있습니다.
위에 언급 했듯이, 여러 파일이 SDF file 내부에 있을 수 있는데요, 어떻게 읽는지 알려드리겠습니다.
from rdkit import Chem
input_sdf = 'input.sdf'
rdmols = Chem.SDMolSupplier(input_sdf)똑같습니다.
여기서 주의 해야할 점은 둘이 똑같다는 것인데요.
둘 다 generator로 나오게 됩니다.
따라서 SDF파일 안에 하나의 화합물만 들어있어도,
rdkit으로 읽게 되면 generator 안에 있기 때문에,
인덱싱을 통해서 rdkit object를 꺼내서 사용하셔야 합니다.
Mol2 File
from rdkit import Chem
input_mol2 = 'input.mol2'
rdmol = Chem.MolFromMol2File(input_mol2)위와 같이 읽을 수 있습니다.
PDB File을 읽는 것은 생략하려고 하는데, 그 이유는 Mol2를 읽는 것과 거의 비슷합니다.
rdkit에서 만들때, 유저의 혼동 및 유지보수를 위해서 사용을 어렵지 않게 해놓은 것 같아요.
마치며
이렇게 rdkit을 설치하고, chemical 파일 포맷과 rdkit을 이용해서 읽는 방법에 대해서 알아봤습니다.
python 덕분에 편하게 사용할 수 있어서 참 다행인 것 같습니다.
rdkit getting started 링크를 참조에 넣어두니 참고 해주세요 !
참조

훌륭한 글이네요. 감사합니다.