#Differntial Expression Gene (DEG) Analysis
Step 1. Performe DEG analysis with DESeq2
이전 글에서 만든 dds 데이터와 DESeq2 함수를 이용하여 DEG 분석을 진행합니다.
DESeq2 분석 결과를 통해 어떤 유전자가 발현량에 있어서 얼마나 그룹 간 차이가 나는 지 확인할 수 있습니다.
예시로, adjusted P value(padj) 값이 낮은 순으로 결과를 정렬하여 확인해보니, Kcng4 라는 유전자가 그룹 간 차이가 가장 높은 것을 볼 수 있습니다.
deseq2.dds <- DESeq(dds)
deseq2.res <- results(deseq2.dds)
deseq2.res <- deseq2.res[order(deseq2.res$padj),]
deseq2.res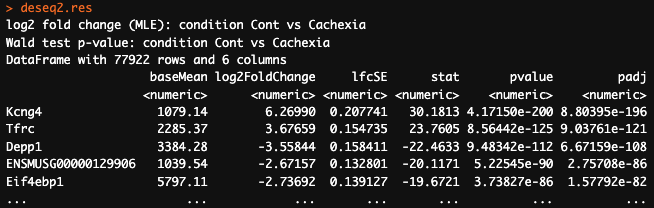
Step 2. Visualization sample-to-sample distances with PCA plot
PCA plot은 주성분(Principal Components)을 이용하여 실험 조건에 따라 샘플의 클러스터링이 잘 되어있는지, 샘플 중 outlier는 없는지 시각적으로 확인할 수 있도록 도와주는 plot입니다.
#Normalization dds data using vst
vsd1 <- vst(deseq2.dds, blind = F)
#Creating PCA plot using normalized data
plotPCA(vsd1, intgroup = 'condition') 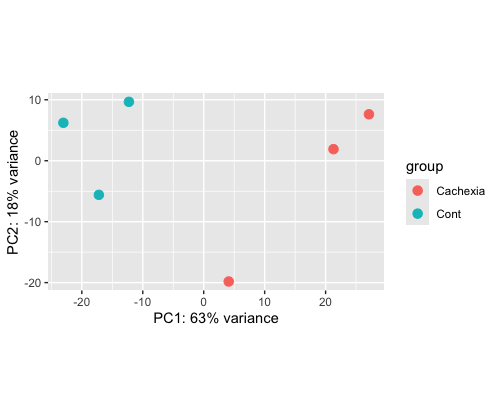
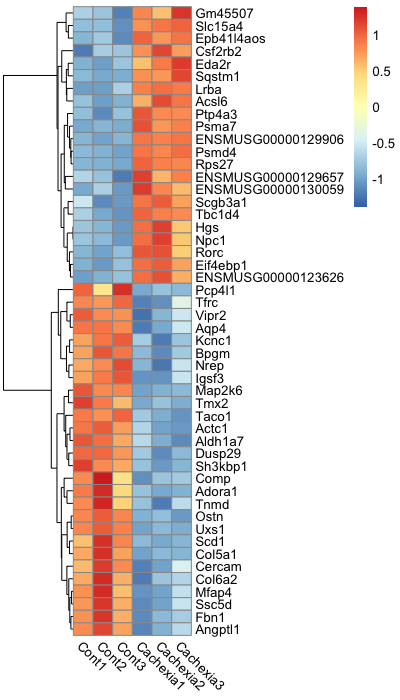
Step 3. Visualization DEGs with heatmap plot
Heatmap plot은 유전자 발현 패턴을 시각화하여 보여주기 때문에 샘플 간 유전자 발현 패턴을 시각적으로 쉽게 비교할 수 있습니다.
조건(Condition)에 따라 유전자의 발현 패턴이 유의하게 다른 top50 유전자의 발현 패턴을 heatmap 상으로 확인하면 다음과 같습니다.
# Creating heatmap
library("pheatmap")
# Select top 50 DEGs
res1 <- deseq2.res
res1 <- na.omit(res1)
res_ordered1 <- res1[order(res1$padj),]
top_genes1 <- row.names(res_ordered1)[1:50]
# Extract counts and normalize
counts1 <- counts(deseq2.dds, normalized = T)
counts_top1 <- counts1[top_genes1,]
log_counts_top1 <- log2(counts_top1 + 1) #log값으로 transformation
new_order <- c("Cont1", "Cont2", "Cont3", "Cachexia1", "Cachexia2", "Cachexia3")
log_counts_top1 <- log_counts_top1[, new_order]
log_counts_top1
pheatmap(log_counts_top1, scale = 'row', cluster_cols=F, angle_col = 315)
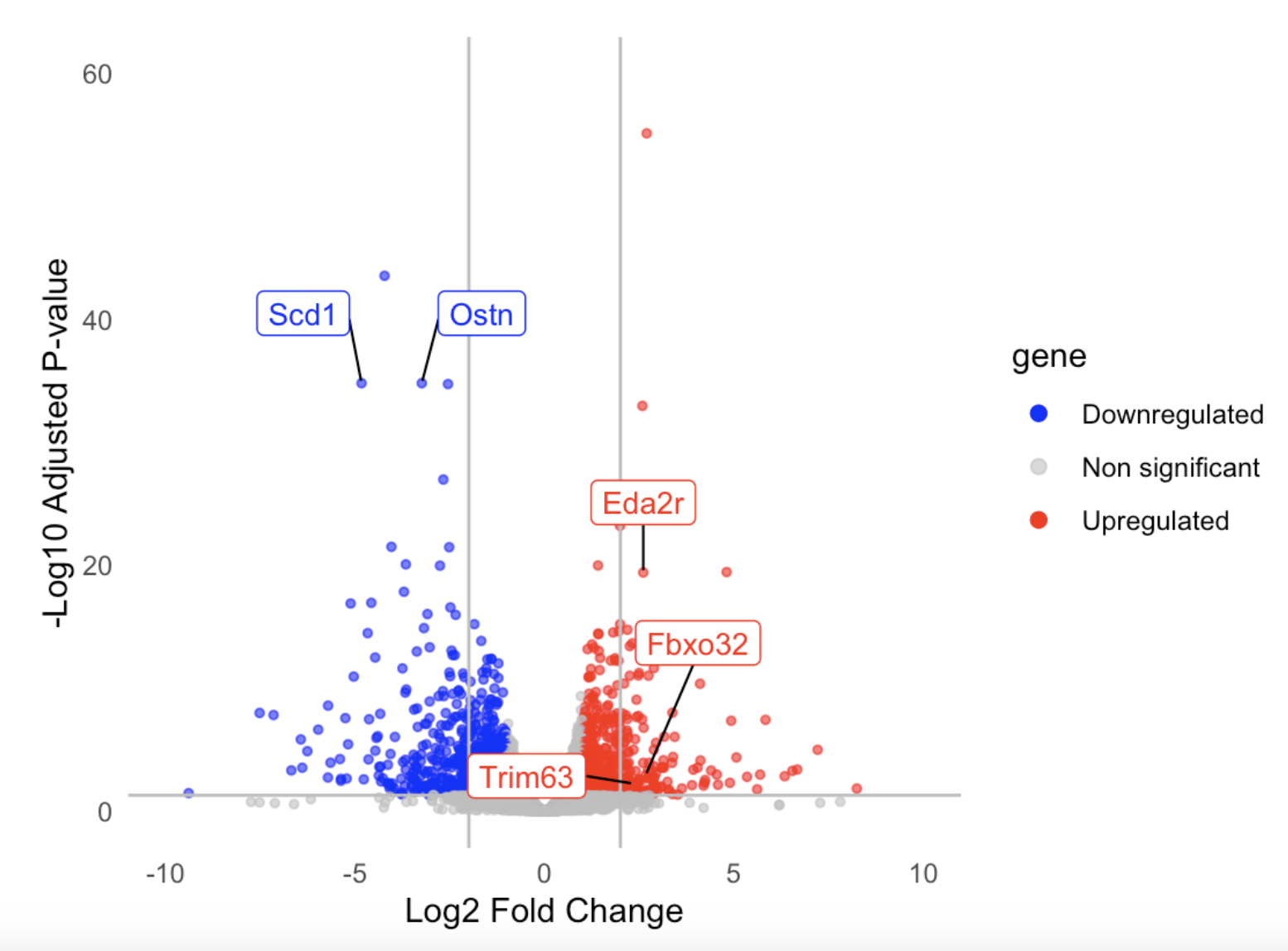
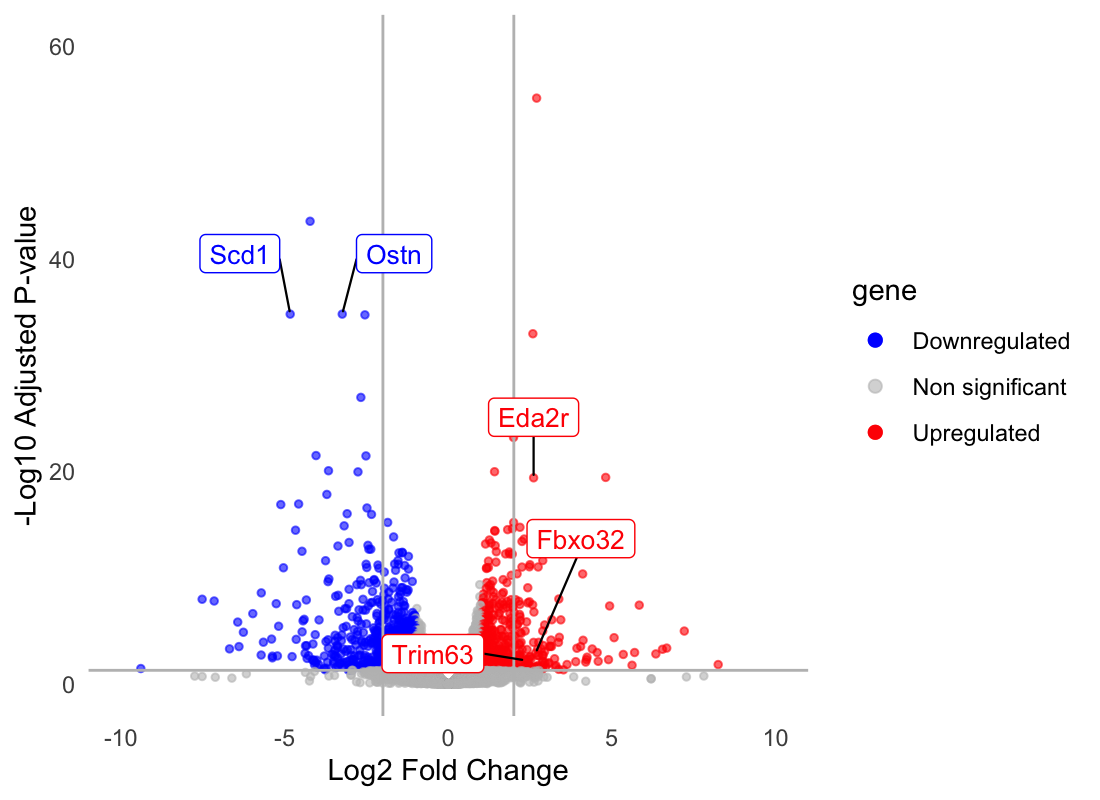
Step 4. Visualization DEGs with Volcano plot
마지막으로, Volcano plot을 이용하면 조건에 따라 어떤 유전자가 많이 발현하고 적게 발현하고 있는지 좀 더 직관적으로 확인할 수 있습니다.
저는 Log2 fold change값이 |2| 이상이면서 padj값이 0.05이하인 유전자들을 효과적으로 보여주기 위해서 회색선을 그어주었습니다.
즉, 회색선 바깥에 있는 유전자들이 Contorl에 비해 유의하게 적게 발현하거나 많이 발현하는 DEG라고 기준을 세운 것입니다.
이러한 기준은 연구자의 주관에 따라 달라질 수 있고, 그에 따라 결과에 대한 해석 또한 달라질 수 있기 때문에, 연구자는 다양한 객관적 근거들을 바탕으로 기준을 설정하는 것이 바람직합니다.
## Volcano plot 그리기
library(ggrepel)
res_df <- as.data.frame(deseq2.res)
res_df <- na.omit(res_df)
res_df$gene <- row.names(res_df)
res_df$gene_symbol <- row.names(res_df)
res_df$log2FoldChange <- -res_df$log2FoldChange
# Create volcano plot
res_df$gene <- "Non significant"
res_df$gene[res_df$log2FoldChange > 1 & res_df$padj < 0.05] <- "Upregulated"
res_df$gene[res_df$log2FoldChange < -1 & res_df$padj < 0.05] <- "Downregulated"
mycolors <- c("Downregulated" = "blue", "Upregulated" = "red", "Non significant" = "grey")
res_df$delabel <- NA
res_df$delabel[res_df$gene != "Non significant"] <- res_df$gene_symbol[res_df$gene != "Non significant"]
annotation <- res_df %>% filter(gene_symbol %in% c("Eda2r", "Scd1", "Ostn", "Trim63", "Fbxo32"))
volcano_plot <- ggplot(res_df, aes(x =log2FoldChange, y = -log10(padj),col = gene, label = delabel)) +
geom_point(alpha = 0.6, size = 1) +
guides(color = guide_legend(override.aes = list(size = 2))) +
scale_color_manual(values = mycolors) +
labs(x = "Log2 Fold Change", y = "-Log10 Adjusted P-value") +
theme_minimal() +
theme(panel.grid.major = element_blank(), panel.grid.minor = element_blank(), legend.position = "right") +
geom_vline(xintercept = c(-2, 2), col = "grey") +
geom_hline(yintercept = -log10(0.05), col = "grey") +
geom_label_repel(
data = annotation,
box.padding = 1,
segment.color = 'black',
segment.size = 0.4,
size = 3.5,
max.overlaps = 20,
key_glyph = draw_key_point,
force = 5,
nudge_y = 5
) +
ylim(c(0,60)) +
xlim(c(-10, 10))
print(volcano_plot)
Supplement. Visualization DEGs with main plot
DEGs를 plot을 통해 보여줄 수 있는 방법은 참 다양합니다.
Log2 Fold Change 값과 normalized counts 값을 사용해서 DEGs가 어떻게 분포되어 있는 지도 나타낼 수 있습니다.
그래서 본인의 데이터를 가장 잘 보여줄 수 있는 형태의 plot이 무엇인지 고민하는 것 또한 중요합니다.
## Main plot 그리기
library(ggplot2)
tmp <- deseq2.res
tmp <- na.omit(tmp)
plot(tmp$baseMean, tmp$log2FoldChange, pch=20, cex=0.45, ylim=c(-3, 3), log="x", col="darkgray", main="DEG Dessication (pval <= 0.05)", xlab="mean of normalized counts", ylab="Log2 Fold Change")
tmp.sig <- tmp[!is.na(tmp$padj) & tmp$padj <= 0.05, ]
points(tmp.sig$baseMean, tmp.sig$log2FoldChange, pch=20, cex=0.45, col="red")
abline(h=c(-1,1), col="blue")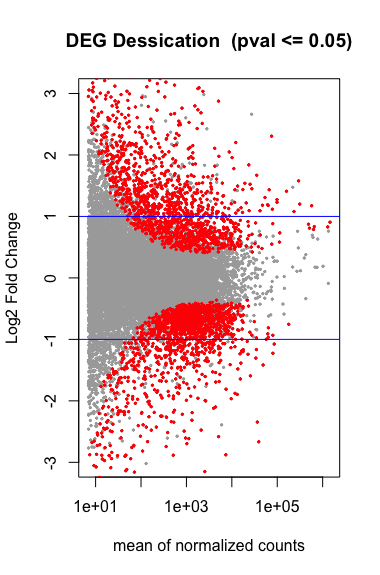
이번 글에서는 실제로 DEGSeq2 함수를 통해 DEGs 데이터를 얻어내고, 데이터를 다양한 Plot을 통해 시각화하는 과정까지 다루었습니다.
다음 글에서는 DEGs가 어떤 생물학적 의미를 갖는지 엿볼 수 있는 다양한 Pathway analysis에 대해서 다루도록 하겠습니다.
감사합니다.
