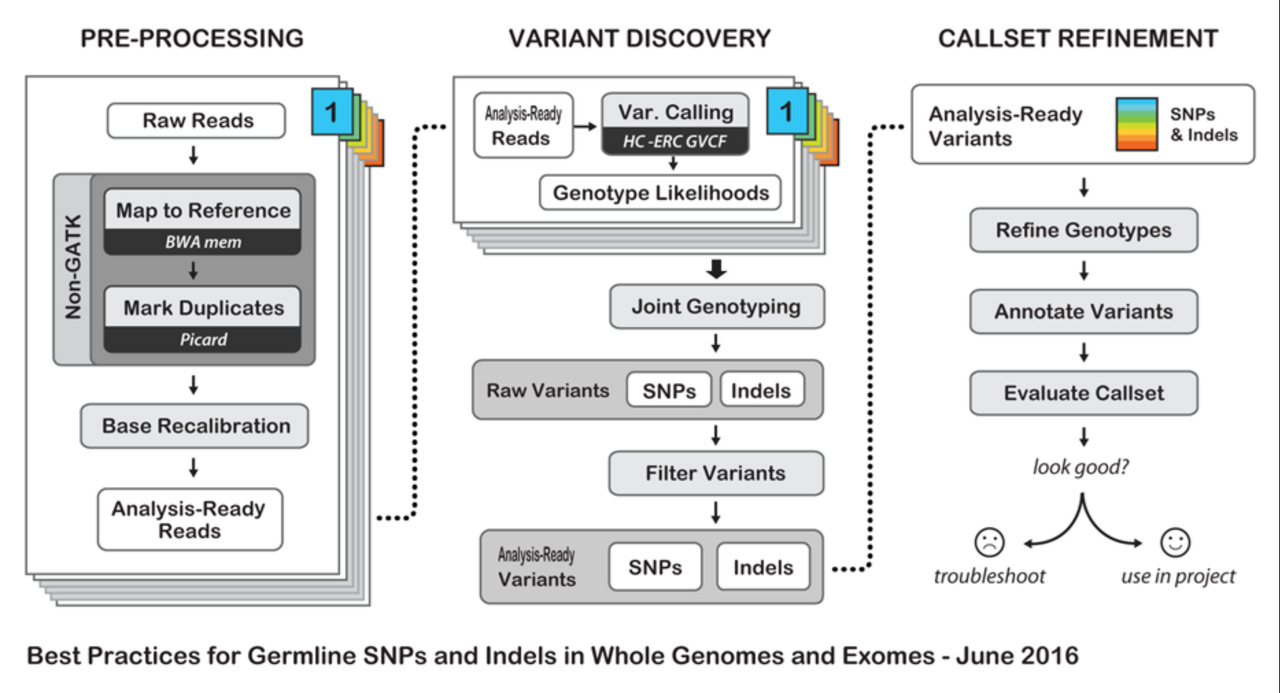
Guideline
할줄아는 언어가 없던 시절 쉘로 무작정 짰던 파이프라인
막상 오류투성이라 오토런이 잘 돌지는 않았고 스텝별로 돌리는게 더 편했지만,,
전체적인 흐름 파악에 좋아서 아직 갖고있는 코드
00. Preparation
Reference genome file
Annotation.vcf file
Sample data
####00_preparation####
#Reference indexing 미리 진행할 것!
- reference genome file
- annotaion.vcf file
※ 업데이트 수시로 확인할것!
#sample data는 미리 백업받아둘 것!
#xargs 명령어 사용시 뒤에 &(백그라운드실행) 추가 하지 말것 --> PID 관리어려움
(1) sample data quality control
(1-1) trimming
** adapter truseq2 : 구버전, truseq3 : 신버전
ex) truseq2 trimming
java -jar /home/kgw/program/Trimmomatic-0.36/trimmomatic-0.36.jar PE -phred33 -threads 4 \
./raw/$1_R1.fastq.gz \
./raw/$1_R2.fastq.gz \
./trim_truseq2/$1_R1_trim_truseq2.paired.fastq.gz ./trim_truseq2/$1_R1_trim_truseq2.unpaired.fastq.gz \
./trim_truseq2/$1_R2_trim_truseq2.paired.fastq.gz ./trim_truseq2/$1_R2_trim_truseq2.unpaired.fastq.gz \
ILLUMINACLIP:/home/kgw/program/Trimmomatic-0.36/adapters/TruSeq2-PE.fa:2:30:10 LEADING:3 TRAILING:3 SLIDINGWINDOW:4:15 MINLEN:36 \
1> ./trim_truseq2/$1.truseq2.log 2> ./trim_truseq2/$1.truseq2.err
(1-2) fastqQC
ex) 4쓰레드 10개씩 병렬
$ ls -1 *fastq* | xargs -P10 -n1 /home/kgw/program/FastQC/fastqc -o ./qc/ -t 4
(2) Reference data indexing
1) reference genome indexing
# genome file은 압축을 해제하고 진행한다. faidx 생성시 gzip 파일 인식을 못하기 때문
$ bwa index -a bwtsw $1.fa 1> bwaindex.err 2> bwaindex.out
$ samtools faidx $1.fa 1> faidx.err 2> faidx.out
$ samtools dict $1.fa -o $1.dict 1> dict.err 2> dict.out
(2) annoation file indexing
# ensembl.org에 이미 .tbi file이 있다면 pass
# 간혹 뒤에 과정에서 오류가 발생하는 경우가 있음 그럴때에는 필요한 부분을 고쳐주고 다시 indexing 진행할 것
$ tabix -p vcf *.vcf.gz01. Bam file 생성
####01_makebamfile####
#sample list file을 만든 후 진행
$ ls -1 Data/* | awk -F "_" '{print $1}' | sort | uniq | cat > data_list
#01_makebam file 실행
#fastq->sam->bam
$ cat data_list | xargs -P10 -n1 -I{} taskset -c 40-59,120-139,60-79,140-159 nohup sh 01_makebam {} 2>&1 /dev/null01_makebam.sh
##############################################
# Set ${RAM} to use for all java processes
RAM=-Xmx20g
#Set number of threads to the number of cores/machine for speed optimization
#nct is num of CPUs, nt is num of walkers(2GB=1walker)
t=8
nt=10
##############################################
# Set the reference fasta
ref=$(ls -1 Ref/*.fa)
#annotaion
ann=$(ls -1 Annotations/*.vcf.gz)
#tmp folder
tmp=tmp/$1
#settingtools
PICARD=/home/Program/picard_2.10.7.jar
GATK=/home/Program/GenomeAnalysisTK_nightly170929.jar
###############################################
# (1) step01_Mapping
# Reference DNA genome sequence 와 분석할 개체의 DNA sequence를 mapping 하는 과정
# 오래걸리는 부분
bwa mem ${ref} \
-t${t} -M \
Data/$1_R1_trim_truseq2.paired.fastq.gz \
Data/$1_R2_trim_truseq2.paired.fastq.gz \
2> Results/$1.bwa.err \
| gzip \
1> Results/$1.sam.gz \
2> Results/$1.sam.gz.err
# (2) step02_Picard
# Bam file 재정리(1)
# 중복제거, Fixmate ...
# 오래걸리는 부분
# 당시 사용하던 서버 기준 각 샘플 파일 당 1시간 30분 소요
# RGSM, RGID is important
# if you want, SILENT --> SILENT
java -jar -Djava.io.tmpdir=${tmp} ${RAM} \
$PICARD AddOrReplaceReadGroups \
INPUT=Results/$1.sam.gz \
OUTPUT=Results/$1.RGsorted.bam \
SORT_ORDER=coordinate \
RGID=$1 \
RGLB=$1LIB \
RGPL=ILLUMINA \
RGPU=NONE \
RGSM=$1 \
VALIDATION_STRINGENCY=SILENT \
2> Results/$1.RGsorted.bam.err
java -jar -Djava.io.tmpdir=${tmp} ${RAM} \
$PICARD MarkDuplicates \
INPUT=Results/$1.RGsorted.bam \
OUTPUT=Results/$1.RGsorted.dedupped.bam \
METRICS_FILE=Results/$1.RGsorted.dedupped.metrics \
REMOVE_DUPLICATES=true \
ASSUME_SORTED=true \
VALIDATION_STRINGENCY=SILENT \
MAX_FILE_HANDLES=1024 \
2> Results/$1.RGsorted.dedupped.bam.err
java -jar -Djava.io.tmpdir=i${tmp} ${RAM} \
$PICARD FixMateInformation \
INPUT=Results/$1.RGsorted.dedupped.bam \
OUTPUT=Results/$1.RGsorted.dedupped.fixmate.bam \
SORT_ORDER=coordinate \
VALIDATION_STRINGENCY=SILENT \
2> Results/$1.RGsorted.dedupped.fixmate.bam.err
# (3) step03_BaseRecallibrator
# Bam file 재정리(2)
# option
#-nt : num of walkers --> multi thread
# requires 2 Gb of memory to run 1 walker
# • 160 --> 320gb 필요 : zeus 최대치
# WARN 00:40:34,859 MicroScheduler - Number of requested GATK threads 400 is more than the number of available processors on this machine 160
#-nct : num of CPUs
java -jar -Djava.io.tmpdir=${tmp} ${RAM} \
$GATK \
-T BaseRecalibrator \
-R ${ref} \
-nct ${t} \
-knownSites ${ann} \
--unsafe \
-I Results/$1.RGsorted.dedupped.fixmate.bam \
-o Results/$1.RGsorted.dedupped.fixmate.recal.grp \
1> Results/$1.RGsorted.dedupped.fixmate.recal.out \
2> Results/$1.RGsorted.dedupped.fixmate.recal.err
java -jar -Djava.io.tmpdir=${tmp} ${RAM} \
$GATK \
-T PrintReads \
-R ${ref} \
-nct ${t} \
--unsafe \
-BQSR Results/$1.RGsorted.dedupped.fixmate.recal.grp \
-I Results/$1.RGsorted.dedupped.fixmate.bam \
-o Results/$1.RGsorted.dedupped.fixmate.recal.bam \
1> Results/$1.RGsorted.dedupped.fixmate.recal.bam.out \
2> Results/$1.RGsorted.dedupped.fixmate.recal.bam.err
# (4-1) step04_IndelRealigner
# Bam file 재정리(3)
# option
#-nt : num of walkers --> multi thread
# • requires 2 Gb of memory to run 1 walker
# • 160 --> 320gb 필요 : zeus 최대치
# WARN 00:40:34,859 MicroScheduler - Number of requested GATK threads 400 is more than the number of available processors on this machine 160
# ※ 두번째 단계에서 지원하지 않음
#-nct : num of CPUs ※ 이 단계에서는 지원하지 않음
# nt 160 : 2시간정도
# nohup 걸면 nohup.out 으로 log 기록함 sh command > /dev/null 2>&1 & 필요
java -jar -Djava.io.tmpdir=${tmp} \
$GATK \
-T RealignerTargetCreator \
-R ${ref} \
-nt ${nt} \
--unsafe \
-I Results/$1.RGsorted.dedupped.fixmate.recal.bam \
-o Results/$1.RGsorted.dedupped.fixmate.recal.intervals \
1> Results/$1.RGsorted.dedupped.fixmate.recal.intervals.out \
2> Results/$1.RGsorted.dedupped.fixmate.recal.intervals.err
java -jar -Djava.io.tmpdir=${tmp} \
$GATK \
-T IndelRealigner \
-R ${ref} \
--unsafe \
-targetIntervals Results/$1.RGsorted.dedupped.fixmate.recal.intervals \
-I Results/$1.RGsorted.dedupped.fixmate.recal.bam \
-o Results/$1.RGsorted.dedupped.fixmate.recal.realigned.bam \
1> Results/$1.RGsorted.dedupped.fixmate.recal.realigned.bam.out \
2> Results/$1.RGsorted.dedupped.fixmate.recal.realigned.bam.err#####RESEQ ERROR LIST####
### bwa에서 제목을 다르게 인식하여 생기는 오류
# ERROR! [mem_sam_pe] paired reads have different names:
# Ex) @SRR1581057.1.1 --> @SRR1581057.1 1
zcat **_1.fastq.gz | sed '/^/,/H/s/.1 / 1 /' | gzip -c > **_1.fixed.fastq.gz
for i in $(ls -1 *_1.fastq.gz) ; do
zcat ${i} | sed '/^/,/ /s/.1 / 1 /' | gzip -c > ${i}.fixed.fastq.gz ; done
for i in $(ls -1 *_2.fastq.gz) ; do
zcat ${i} | sed '/^/,/ /s/.2 / 2 /' | gzip -c > ${i}.fixed.fastq.gz ; done
### Annotation file에 empty allele이 존재할 경우
awk -F '\t' '($1 ~ /##/)' **.vcf > **_#.vcf
awk -F '\t' '{if (length($5) != 0) print }' **.vcf > **.delempty.vcf
cat Bos_taurus_#.vcf Bos_taurus.delempty.vcf > Bos_taurus.fix.vcf
# more -10 Bos_taurus.blank.vcf 로 확인할것!
# --> bgzip Bos_taurus.fix.vcf
# --> tabix -p vcf Bos_taurus.fix.vcf.gz
### Annotation file에 field value값에 space가 있는 경우
# Ex) Axiom Genotyping Array_1;TSA=SNV;Reference_seq=A;Variant_seq=G" --> AxiomGenotypingArray
awk -F '\t' '{gsub(/ /,"",$0);print}' Sus_scrofa.vcf > Sus_scrofa.cleaned.vcf
# --> bgzip Sus_scrofa.cleaned.vcf
# --> tabix -p vcf Sus_scrofa.cleaned.vcf.gz
### Annotation 인덱싱 여부 : snp indexing
nohup tabix -p vcf Bos_taurus.clean.vcf.gz &
### 템프디렉토리에 여러 실행파일이 한번에 들어가면 용량 과다로 작업 안됨
#- tmp dir 설정
-Djava.io.tmpdir=./tmp1
### 64비트는 메모리 사용 무한대로 씀
#- 용량제한
> -Xmx2g
### empty allele 오류
#- remove blank allele
awk -F '\t' '($1 ~ /##/)' Bos_taurus.vcf > Bos_taurus_#.vcf
awk -F '\t' '{if (length($5) != 0) print }' Bos_taurus.vcf > Bos_taurus.delempty.vcf
cat Bos_taurus_#.vcf Bos_taurus.delempty.vcf > Bos_taurus.fix.vcf
more -10 Bos_taurus.blank.vcf 로 확인할것!
bgzip Bos_taurus.fix.vcf
tabix -p vcf Bos_taurus.fix.vcf.gz
### The provided VCF file is malformed at approximately line number 2036: The VCF specification does not allow for whitespace in the INFO field. Offending field value was "Axiom Genotyping Array_1;TSA=SNV;Reference_seq=A;Variant_seq=G", for input source: /BiO/home/kgw/pig_sra_20170731/test/kwb/annotations/Sus_scrofa.fix.vcf.gz
#- Axiom Genotyping Array —> 띄어쓰기 때문에 생긴 오류
# —> 띄어쓰기 제거
awk -F '\t' '{gsub(/ /,"",$0);print}' Sus_scrofa.vcf > Sus_scrofa.cleaned.vcf
bgzip Sus_scrofa.cleaned.vcf
tabix -p vcf Sus_scrofa.cleaned.vcf.gz
### Phred Score Error
##### ERROR MESSAGE: SAM/BAM/CRAM file htsjdk.samtools.SamReader$PrimitiveSamReaderToSamReaderAdapter@813ab53 appears to be using the wrong encoding for quality scores: we encountered an extremely high quality score of 62. Please see [https://software.broadinstitute.org/gatk/documentation/article?id=6470](https://software.broadinstitute.org/gatk/documentation/article?id=6470) for more details and options related to this error.
'''
#step03_BaseRecallibrator_FixQualityScores
java -jar -Djava.io.tmpdir=${tmp} ${RAM} \
$GATK \
-T BaseRecalibrator \
-R ${ref} \
-nct ${t} \
-knownSites ${ann} \
--unsafe \
--fix_misencoded_quality_scores \ ### add and run again
-I Results/$1.RGsorted.dedupped.fixmate.bam \
-o Results/$1.RGsorted.dedupped.fixmate.recal_fixqualscore.grp \
1> Results/$1.RGsorted.dedupped.fixmate.recal_fixqualscore.out \
2> Results/$1.RGsorted.dedupped.fixmate.recal_fixqualscore.err
java -jar -Djava.io.tmpdir=${tmp} ${RAM} \
$GATK \
-T PrintReads \
-R ${ref} \
-nct ${t} \
--unsafe \
-BQSR Results/$1.RGsorted.dedupped.fixmate.recal_fixqualscore.grp \
-I Results/$1.RGsorted.dedupped.fixmate.bam \
-o Results/$1.RGsorted.dedupped.fixmate.recal.bam \
1> Results/$1.RGsorted.dedupped.fixmate.recal.bam.out \
2> Results/$1.RGsorted.dedupped.fixmate.recal.bam.err
```01_2.1. Chromosome 분리
# (4-2) step04_SplitBAM
# (1) 원본 index, picard 통과했으면 필요없는 부분
samtools index
# (2) 분리
for chrom in seq 1 18 X Y MT
do
samtools view -bh $1 chr${chrom}
Done
# 주의
# chr1로 인식하는 경우도 있고 그냥 1만 써야 인식하는 경우도 있음 --> 샘플마다 다름!
samtools view P06073F1.fixed.RGsorted.bam 21 -o P06073F1_chr21.bam &
samtools view P06073F1.fixed.RGsorted.bam 22 -o P06073F1_chr22.bam &
samtools view P06073F1.fixed.RGsorted.bam 23 -o P06073F1_chr23.bam &
samtools view P06073F1.fixed.RGsorted.bam 24 -o P06073F1_chr24.bam &
samtools view P06073F1.fixed.RGsorted.bam 25 -o P06073F1_chr25.bam &
1. sort
samtools sort chr${chrom}
2. index
samtools index chr${chrom}.bam01_2.2. Scaffold 분리가 필요할 때
### scaffold 분리
samtools view -h $1 | awk '{FS="\t";OFS="\t"}{if($1 ~ "@" || $3 != "1" && $3 != "2" && $3 != "3" && $3 != "4" && $3 != "5" && $3 != "6" && $3 != "7" && $3 != "8" && $3 != "9" && $3 != "10" && $3 != "11" && $3 != "12" && $3 != "13" && $3 != "14" && $3 != "15" && $3 != "16" && $3 != "17" && $3 != "18" && $3 != "19" && $3 != "20" && $3 != "21" && $3 != "22" && $3 != "23" && $3 != "24" && $3 != "25" && $3 != "26" && $3 != "27" && $3 != "28" && $3 != "29" && $3 != "30" && $3 != "31" && $3 != "X" && $3 != "Y" && $3 != "MT"){print $0}}' | cat > sample_scaffold.RGsorted.bam &
Samtools sort -o $1.scaffold.sorted.bam $1.scaffold.bam
#한번에 진행ver -돼지
samtools view -h $1 | awk '{FS="\t";OFS="\t"}{if($1 ~ "@" || $3 != "1" && $3 != "2" && $3 != "3" && $3 != "4" && $3 != "5" && $3 != "6" && $3 != "7" && $3 != "8" && $3 != "9" && $3 != "10" && $3 != "11" && $3 != "12" && $3 != "13" && $3 != "14" && $3 != "15" && $3 != "16" && $3 != "17" && $3 != "18" && $3 != "X" && $3 != "Y" && $3 != "MT"){print $0}}' | samtools sort -o ./bam/BKS02_scaffold2.bam &
# 닭
samtools view -h Results/$1.RGsorted.dedupped.fixmate.recal.realigned.bam | awk '{FS="\t";OFS="\t"}{if($1 ~ "@" || $3 != "1" && $3 != "2" && $3 != "3" && $3 != "4" && $3 != "5" && $3 != "6" && $3 != "7" && $3 != "8" && $3 != "9" && $3 != "10" && $3 != "11" && $3 != "12" && $3 != "13" && $3 != "14" && $3 != "15" && $3 != "16" && $3 != "17" && $3 != "18" && $3 != "19" && $3 != "20" && $3 != "21" && $3 != "22" && $3 != "23" && $3 != "24" && $3 != "25" && $3 != "26" && $3 != "27" && $3 != "28" && $3 != "30" && $3 != "31" && $3 != "32" && $3 != "33" && $3 != "W" && $3 != "Z" && $3 != "MT"){print $0}}' | samtools sort -o ./Results/chr/scaffolds/$1.scaffolds.bam
Samtools index $1.scaffold.bam02. GATK Haplotypecaller - GVCF 생성
- 실제 mapping 을 하는 부분
####02_makeVCFfile####
#chr file 생성
$ grep ">" Ref/*.fa | grep -B 100 MT | awk -F " " '{print $1}' | sed 's/>//' | cat > chr
#chr folder 생성
$ mkdir Results/chr
$ for i in $(cat chr); do mkdir Results/chr/${i}; done
#실행
$ cat data_list | xargs -P10 -n1 -I{} taskset -c 40-59,120-139,60-79,140-159 nohup sh 02_makevcf {} 2>&1 /dev/null
#scaffold는 -L 옵션을 -XL 옵션으로 바꿔주고 옵션에 chr file을 넣어준다02_makevcf.sh
##############################################
# Set ${RAM} to use for all java processes
RAM=-Xmx20g
#Set number of threads to the number of cores/machine for speed optimization
#nct is num of CPUs, nt is num of walkers(2GB=1walker)
t=8
nt=10
##############################################
# Set the reference fasta
ref=$(ls -1 ./Ref/*.fa)
#annotaion
ann=$(ls -1 ./Annotations/*.vcf.gz)
#tmp folder
tmp=./tmp/$1
#settingtools
PICARD=/home/Program/picard_2.10.7.jar
GATK=/home/Program/GenomeAnalysisTK_nightly170929.jar
###############################################
# step05_0_makeChrfileFolder
#chr file 생성
grep ">" Ref/*.fa | grep -B 100 MT | awk -F " " '{print $1}' | sed 's/>//' | cat > chr
#chr folder 생성
mkdir Results/chr
for i in $(cat chr); do mkdir Results/chr/${i}; done
# step05_1_Haplotypecaller
for i in $(cat chr) ; do
java -jar -Djava.io.tmpdir=${tmp} ${RAM} \
${GATK} \
-T HaplotypeCaller \
-R ${ref} \
-nct ${t} \
-rf BadCigar \
--emitRefConfidence GVCF \
--variant_index_type LINEAR \
--variant_index_parameter 128000 \
--dbsnp ${ann} \
# scaffold는 -L 옵션을 -XL 옵션으로 바꿔주고 옵션에 chr file을 넣어준다
-L $i \
-I Results/$1.RGsorted.dedupped.fixmate.recal.realigned.bam \
-o Results/chr/$i/$1_$i.RGsorted.dedupped.fixmate.recal.realigned.HaplotypeCaller.g.vcf \
1> Results/chr/$i/$1_$i.RGsorted.dedupped.fixmate.recal.realigned.HaplotypeCaller.out \
2> Results/chr/$i/$1_$i.RGsorted.dedupped.fixmate.recal.realigned.HaplotypeCaller.err
bgzip Results/chr/$i/$1_$i.RGsorted.dedupped.fixmate.recal.realigned.HaplotypeCaller.g.vcf
tabix -p vcf Results/chr/$i/$1_$i.RGsorted.dedupped.fixmate.recal.realigned.HaplotypeCaller.g.vcf.gz ; done03. Variation file 생성**
####03_completefile####
#variation file 생성
$ for i in $(ls -1 Results/chr/); do ls -1 ./Results/chr/$i/*vcf.gz | sed -e 's/^/\t--variant /' | cat > ./Results/chr/$i/$i; done
#genome_ver 변수 설정 후 진행
#genome_ver 확인법
# snpEff 폴더를 진행중인 분석폴더의 하위폴더에 위치시킨 후
$ grep 축종의학명의일부 snpEffx.x/snpEff.config
$ cat data_list | xargs -P10 -n1 -I{} taskset -c 40-59,120-139,60-79,140-159 nohup sh 03_completefile {} 2>&1 /dev/null03_completefile.sh
###____setting_file_name____###
#SAMPLE_NAME=$2
##############################################
# Set ${RAM} to use for all java processes
RAM=-Xmx20g
#Set number of threads to the number of cores/machine for speed optimization
#nct is num of CPUs, nt is num of walkers(2GB=1walker)
t=8
nt=10
##############################################
# Set the reference fasta
ref=$(ls -1 ./Ref/*.fa)
#annotaion
ann=$(ls -1 ./Annotations/*.vcf.gz)
#tmp folder
tmp=./tmp/$1
#settingtools
PICARD=/home/Program/picard_2.10.7.jar
GATK=/home/Program/GenomeAnalysisTK_nightly170929.jar
###############################################
###_____snpEff_setting______###
SNPEFFdir=/BiO/home/kgw/program/snpEff4.3/
####____manualy_setting_____###
#genome_ver=$3
###############################
# Chromosome을 쪼개서 프로그램을 돌리기 때문에 var에 넣어줄 파일을 미리 생성해야함
# step06_CombineGVCFs_GenotypeGVCFs
'''
for i in $(ls -1 Results/chr/); do ls -1 ./Results/chr/$i/*vcf.gz | sed -e 's/^/\t--variant /' | cat > ./Results/chr/$i/$i; done
'''
var=$(cat ./Results/chr/$1/$1)
# step07_VariantFiltration
java -jar \
$GATK \
-T VariantFiltration \
-R ${ref} \
-V Results/${SAMPLE_NAME}_$1.RGsorted.dedup.recal.realigned.g.vcf.gz \
-o Results/${SAMPLE_NAME}_$1.RGsorted.dedup.recal.realigned.filtered.vcf \
--filterName LowReadPosRankSum --filterExpression "ReadPosRankSum < -2.0" \
--filterName LowMQRankSum --filterExpression "MQRankSum < -2.0" \
--filterName LowQual --filterExpression "QUAL < 30.0" \
--filterName QD --filterExpression "QD < 3.0" \
--filterName FS --filterExpression "FS > 30.0" \
--filterName MQ --filterExpression "MQ < 30.0" \
--filterName DP --filterExpression "DP < 7" \
--genotypeFilterName DP --genotypeFilterExpression "DP < 7" \
--genotypeFilterName GQ --genotypeFilterExpression "GQ < 10.0" \
--missingValuesInExpressionsShouldEvaluateAsFailing \
2> Results/${SAMPLE_NAME}_$1.RGsorted.dedup.recal.realigned.filtered.vcf.err
# step08_SelectVariants
java -jar \
$GATK \
-T SelectVariants \
-R ${ref} \
-V Results/${SAMPLE_NAME}_$1.RGsorted.dedup.recal.realigned.filtered.vcf \
-o Results/${SAMPLE_NAME}_$1.RGsorted.dedup.recal.realigned.filtered.selected.vcf \
--excludeFiltered \
--excludeNonVariants \
--setFilteredGtToNocall \
# --selectTypeToInclude $4 \ ### SNP/INDEL 중 하나만 뽑을 때 사용
2> Results/${SAMPLE_NAME}_$1.RGsorted.dedup.recal.realigned.filtered.selected.vcf.err
# step09_snpEff
java -jar ${RAM} \
${SNPEFFdir}/snpEff.jar \
-c ${SNPEFFdir}/snpEff.config \
-s Results/${SAMPLE_NAME}_$1.RGsorted.dedup.recal.realigned.snpEff.summary.html \
-csvStats Results/${SAMPLE_NAME}_$1.RGsorted.dedup.recal.realigned.snpEff.summary.csv \
-strict \
$3 \
Results/${SAMPLE_NAME}_$1.RGsorted.dedup.recal.realigned.filtered.selected.vcf \
1> Results/${SAMPLE_NAME}_$1.RGsorted.dedup.recal.realigned.filtered.snpEff.vcf \
2> Results/${SAMPLE_NAME}_$1.RGsorted.dedup.recal.realigned.filtered.snpEff.vcf.err04. optional
ExtractFields
# step10_snpsift_extractFields
cat results/Bos_taurus.RGsorted.dedup.recal.realigned.filtered.selected.snpEff.vcf | \
perl /home/disk_add/disk3/01kgw/NBIT/bin/snpEff_core_4.2_20151205/scripts/vcfEffOnePerLine.pl | \
/home/disk_add/disk3/01kgw/NBIT/bin/jre1.8.0_73/bin/java -Xmx2g \
-jar /home/disk_add/disk3/01kgw/NBIT//bin/snpEff_core_4.2_20151205/SnpSift.jar \
extractFields - \
CHROM \
POS \
ID \
REF \
ALT \
FILTER \
"GEN[*].GT" \
"GEN[*].AD" \
"GEN[*].DP" \
"ANN[*].ALLELE" \
"ANN[*].EFFECT" \
"ANN[*].IMPACT" \
"ANN[*].GENE" \
"ANN[*].GENEID" \
"ANN[*].FEATURE" \
"ANN[*].FEATUREID" \
"ANN[*].BIOTYPE" \
"ANN[*].RANK" \
"ANN[*].HGVS_C" \
"ANN[*].HGVS_P" \
"ANN[*].CDNA_POS" \
"ANN[*].CDNA_LEN" \
"ANN[*].CDS_POS" \
"ANN[*].CDS_LEN" \
"ANN[*].AA_POS" \
"ANN[*].AA_LEN" \
"ANN[*].DISTANCE" \
"ANN[*].ERRORS" \
"EFF[*].EFFECT" \
"EFF[*].IMPACT" \
"EFF[*].FUNCLASS" \
"EFF[*].CODON" \
"EFF[*].AA" \
"EFF[*].AA_LEN" \
"EFF[*].GENE" \
"EFF[*].BIOTYPE" \
"EFF[*].CODING" \
"EFF[*].TRID" \
"EFF[*].RANK" \
"LOF[*].GENE" \
"LOF[*].GENEID" \
"LOF[*].NUMTR" \
"LOF[*].PERC" \
"NMD[*].GENE" \
"NMD[*].GENEID" \
"NMD[*].NUMTR" \
"NMD[*].PERC" \
> results/Bos_taurus.RGsorted.dedup.recal.realigned.filtered.snpEff.selected.test.txt
# MISSENSE
cat Pig106.20180305_3_SNP.vcf | perl /home/kgw/program/snpEff4.3/scripts/vcfEffOnePerLine.pl | java -Xmx8g -jar /home/kgw/program/snpEff4.3/SnpSift.jar filter "(ANN[*].EFFECT = 'missense_variant')"FixHeader
python step11_fixHeader.py \
results/Bos_taurus.RGsorted.dedup.recal.realigned.filtered.selected.vcf \
results/Bos_taurus.RGsorted.dedup.recal.realigned.filtered.selected.snpEff.txt \
results/Bos_taurus.RGsorted.dedup.recal.realigned.filtered.selected.snpEff.fixHeader.txtstep01_fixHeader.py
#!/usr/bin/env python
import sys, os, string, operator, glob, gzip, math, time
args = sys.argv
if len(args) != 4:
print 'Usage:', args[0], '.snpEff.vcf .SnpSift.txt .SnpSift.header.txt'
sys.exit()
snpEffname = args[1]
SnpSiftname = args[2]
SnpSiftheaderfix = args[3]
sampleheaders = ''
for lines in open(snpEffname, 'r'):
if lines.startswith('#CHROM'):
sampleheaders = lines
break
samplelist = sampleheaders.rstrip('\n').rstrip('\r').split('\t')[9:]
infile = open(SnpSiftname, 'r')
line1 = infile.readline()
linelist1 = line1.rstrip('\n').rstrip('\r').split('\t')
newlist = linelist1[:6] + [ix+'.GT' for ix in samplelist] \
+ [ix+'.AD' for ix in samplelist] + [ix+'.DP' for ix in samplelist] \
+ [ix.replace('[*]', '') for ix in linelist1[9:]]
outfile = open(SnpSiftheaderfix, 'w')
outfile.write('\t'.join(newlist) + '\n')
while 1:
lines = infile.readline()
if lines == '': break
outfile.write(lines)
outfile.close()Statistics
#!/usr/bin/env python
import sys, os, string, operator, glob, gzip, math, time
args = sys.argv
if len(args) != 2:
print 'Usage:', args[0], '.txt.gz'
sys.exit()
infilename = args[1]
if infilename.endswith('.txt.gz'):
outfilename = infilename.replace('.txt.gz', '') + '.stat2.txt'
elif infilename.endswith('.txt'):
outfilename = infilename.replace('.txt', '') + '.stat2.txt'
else:
sys.exit('wrong input filename')
if infilename.endswith('.gz'):
infile = gzip.GzipFile(infilename, 'r')
else:
infile = open(infilename, 'r')
line1 = infile.readline()
linelist1 = line1.rstrip('\n').split('\t')
samplelist = [linelist1[ix].replace('.GT', '').strip() for ix in range(len(linelist1)) if linelist1[ix].endswith('.GT')]
indexlist = [ix for ix in range(len(linelist1)) if linelist1[ix].endswith('.GT')]
iEffect = linelist1.index('EFF[*].EFFECT')
statdict = {}
_tdict = {}
genotypedict = {}
#icount = 0
chrtmp = ''
while 1:
lines = infile.readline()
if lines == '': break
linelist = lines.rstrip('\n').split('\t')
CHROM, POS, ID, REF, ALT = linelist[:5]
chrkey = CHROM + '\t' + POS + '\t' + REF + '\t' + ALT
genolist = [linelist[ix] for ix in indexlist]
if CHROM != chrtmp: print >> sys.stderr, 'processing...', CHROM
snpefftypes = linelist[iEffect]
snpefftypesSet = set()
for _ix in snpefftypes.split('|'):
for _ij in _ix.split('&'):
if len(_ij) == 0: continue
snpefftypesSet.add(_ij)
for _snpefftype in snpefftypesSet:
_tdict[_snpefftype] = 0
for sampleid, igeno in zip(samplelist, genolist):
if igeno in ('0|0', '0/0', './.'): continue
statdict.setdefault(_snpefftype, {}).setdefault(sampleid, {}).setdefault(igeno, {})
statdict[_snpefftype][sampleid][igeno][chrkey] = 0
statdict.setdefault(_snpefftype, {}).setdefault(igeno, {}) # ALL
statdict[_snpefftype][igeno][chrkey] = 0 # ALL
genotypedict[igeno] = 0
chrtmp = CHROM
#icount += 1
#if icount == 5000: break
infile.close()
genotypes = genotypedict.keys()
genotypes.sort()
snpefftypelist = _tdict.keys()
snpefftypelist.sort()
outfile = open(outfilename, 'w')
plist = []
for sampleid in samplelist:
for _geno in genotypes:
plist.append(sampleid + '_' + _geno)
plist.append(sampleid + '_TOT')
for _geno in genotypes: # ALL
plist.append('ALL_' + _geno) # ALL
plist.append('ALL' + '_TOT') # ALL
wlist = ['snpEff_Effect'] + plist
outfile.write('\t'.join(wlist) + '\n')
for snpefftype in snpefftypelist:
countlist = []
for sampleid in samplelist:
_countlist = []
for _geno in genotypes:
try:
ic1 = len(statdict[snpefftype][sampleid][_geno])
except:
ic1 = 0
_countlist.append(ic1)
totcount = reduce(operator.add, _countlist)
for _iix in _countlist: countlist.append(_iix)
countlist.append(totcount)
_countlist = [] # ALL
for _geno in genotypes: # ALL
try: # ALL
ic1 = len(statdict[snpefftype][_geno]) # ALL
except: # ALL
ic1 = 0 # ALL
_countlist.append(ic1) # ALL
totcount = reduce(operator.add, _countlist) # ALL
for _iix in _countlist: countlist.append(_iix) # ALL
countlist.append(totcount) # ALL
wlist = [snpefftype] + countlist
outfile.write('\t'.join(map(str, wlist)) + '\n')
outfile.close()
"""
740 EFF.EFFECT
747 EFF.BIOTYPE
[deepreds@soy bak.snpEff]$ zcat cohort239.RGsorted.dedup.recal.realigned.split.filtered.snpEff.SnpSift.fixHeader.INDEL.txt.gz | cut -f740,747 |more
"""VCF Extraction
# 분석에 이용할 샘플만 추출할 때 사용
cat *.snpEff.vcf | vcf-subset -c $group > $sample.$group.vcf05. SNP QC
vcftools --vcf $1_SNP.RGsorted.dedup.recal.realigned.filtered.snpEff.vcf --max-missing 0.01 --maf 0.05 --hwe 1e-6 --recode --recode-INFO-all --out $1 2>$1.err
# --keep filename #해당샘플만 추출
내 지식의 외장하드