코로나 바이러스의 변이 우세종을 예측하는 인공지능 개발
위 주제로 졸업 프로젝트를 진행하고 있다.
우선 배경 지식을 먼저 조사하고 멘토링 받아서 정리해보았다.
바이러스의 변이에는 많은 요인들이 작용한다. 그리고 변이가 일어난다고 해서 그것이 반드시 살아남는다는 법은 없다.
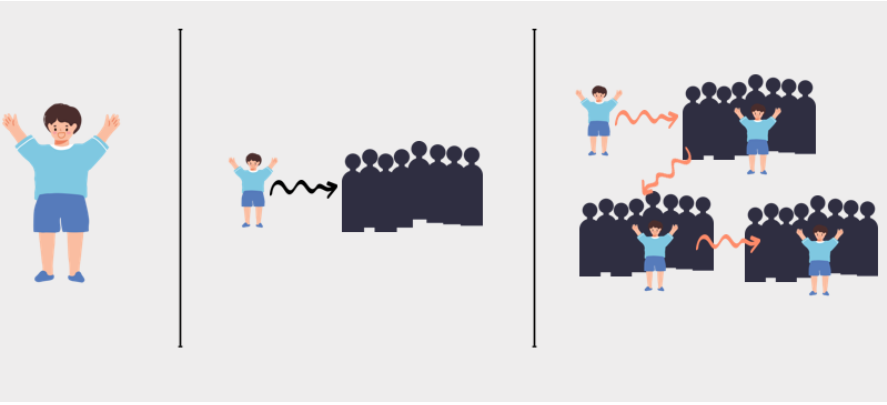
위 그림은 바이러스가 퍼지는 팬데믹 상황을 가장 단순한 모델로 도식화한 것이다. 여러 가지 요인이 작용하고 한 사람에게 감염되는 바이러스도 한 종류는 아니겠지만 일단은 가장 메인인 바이러스 하나에 감염이 된다고 가정을 하고 한 사람에서 여러 사람 -> 여러 사람 중 한 사람에서 또 여러 사람으로 이어지는 감염 과정을 그려보았다.
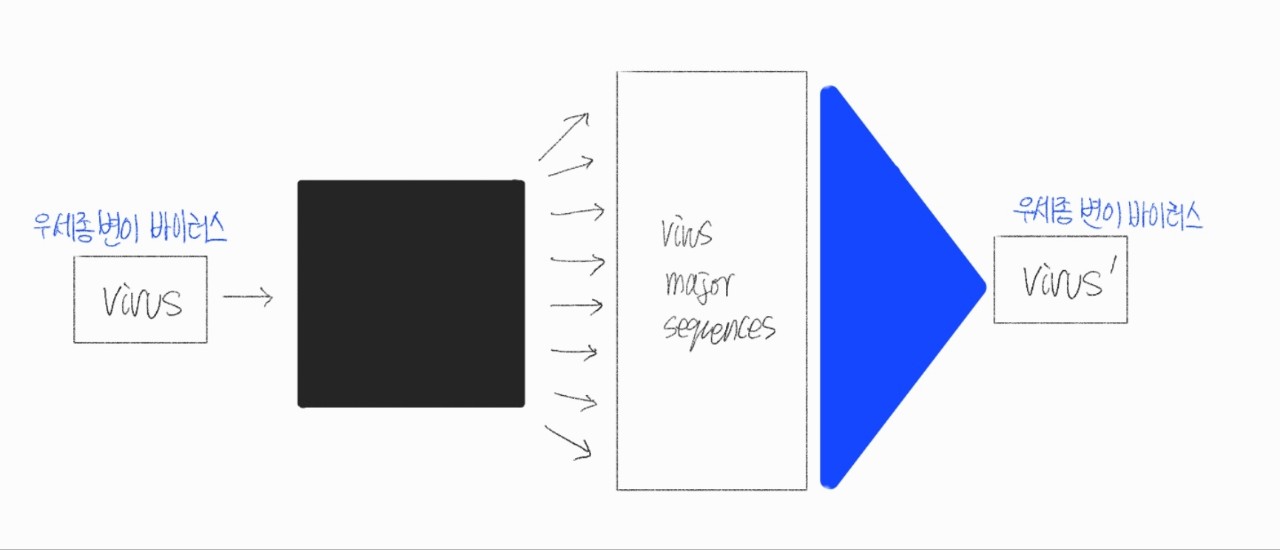
기술을 적용하기에 가장 알맞은 형태의 모델을 도출하는 작업을 했다. 텍스트 생성 모델을 이용해 생존 가능한 바이러스의 변이 염기 서열을 생성한 후 그들 중 가장 전파력이 높을 것으로 예측되는 것이 다음 세대의 우세종이 될 것이라고 예측하는 방식이다.
그런데 이 모델대로 개발을 하려면 텍스트 생성 모델에 학습을 시키기 위한 데이터셋에 전파력을 수치적으로 측정한 데이터가 필요한데 전파력을 수치적으로 계산한다는게 생물학적으로 거의 불가능하다고 한다. 게다가 GAN 모델을 사용하려고 했더니 GAN 모델은 이미지 정보에 대해서는 압도적으로 좋은 성능을 지니지만 텍스트에 관해서는 RNN, LSTM을 이기지 못 한다고 한다. 덤으로 RNN, LSTM은 자연어...이기 때문에 유전자 염기 서열이라는 특수성을 가진 데이터에 대해서는 성능이 입증되지 않았다는 문제가 있었다.
그래서 프로젝트 방향을 어느 정도 선회하기로 했다...
일단 그 전에 바이오인포매틱스 분야에서 염기 서열을 어떻게 처리해서 분석하고 있는지를 알아보기 위해서 몇 가지 논문을 찾아봤다.
https://koreascience.kr/article/CFKO202125036644410.pdf
1. BERT Mask-Filling과 단백질-단백질 상호작용 모델을 이용한 SARS-CoV-2 변이 예측 알고리즘
이건 한국컴퓨터정보학회 하계학술대회에 실린 2p짜리 작은 논문..인데 바이러스의 변이를 예측하기 위한 방법론을 제시하는 정도이고 실제로 이 알고리즘을 개발해서 실행해본 것 같지는 않았다.
1) 단백질 BERT 모델(106M개 이상의 단백질 서열로 사전 학습된 Bidirectional Encoder Representations from Transformers(BERT))을 이용해서 SARS-CoV-2 스파이크 단백질 서열의 임의의 아미노산 잔기를 [MASK] 토큰으로 치환하고 이를 예측시키는 방법으로 단백질에 구조적으로 가능한 돌연변이를 발생시킨다.
2) 단백질-단백질 상호작용 예측 모델(Deep Perpose PPI): 두 단백질을 입력으로 받아 두 단백질이 결합하는지를 예측한다. → 변이가 유도된 SARS-CoV-2 스파이크 단백질의 human ACE2 수용체 결합력을 계산함으로써, 해당 돌연변이가 자연선택에 의해 유전자 풀에서 살아남을지를 예측할 수 있다.
위의 두 방법을 이용하는 것이 주요 아이디어였다. random하게 변이를 일으킨 염기 서열의 단백질 서열과 SARS-Cov-2 바이러스가 인간 세포에 감염될 때 달라붙는 ACE2라는 수용체의 단백질 서열을 Deep Perpose PPI에 입력으로 넣어 우세종 가능성 여부를 판단하겠다는 것이다.
이 방법론에 대해서 생각을 해봤는데 조사해 본 바로는 코로나 바이러스에서 가장 변이가 많이 일어나고 전파력에 큰 영향을 미치는 부분인 spike 단백질의 염기 서열이 약 4100개의 문자열이고 이걸 단백질 서열로 변환하면 약 1320개의 문자열이 된다. 그리고 단백질 서열의 한 자리에 들어갈 수 있는 아미노산의 종류는 20개이니 random 변이는 총 20의 1320제곱의 가지 수를 가진다. 이런 식으로는 계산을 아무리 해도 실제 변이를 예측하기는 어려울 것이다.
https://www.medrxiv.org/content/10.1101/2021.09.07.21263228v1
2. 미국의 공유 차량 서비스 기업 Uber와 하버드 의대가 협업 연구를 한 PyR0
1번 논문의 내용에서 한계를 보완하기 위해서 GAN이든 RNN, LSTM이든 이용해보려고 한 건데 결국 데이터의 특성 때문에 기존 모델들을 학습시키는데 한계가 있다는 것을 알게 되었으니 아예 다른 방법으로 프로젝트를 진행하자고 얘기했다. 그리고 우리가 사용하려는 데이터의 특성에 대해서 아직 정확하게 파악하지 못했다는 생각을 하게 되었고 그에 따라 데이터 분석이 잘 된 예시를 찾아서 공부해보는 것이 프로젝트 주제 선회의 좋은 참고가 될 것이라 여겨 이 논문을 찾아 조사하게 되었다. 1번의 논문과 달리 대형 프로젝트로 실제 진행되었으며 데이터 분석을 통해 여러 차트가 논문에 나와있고 이 차트들을 통해 데이터 분석 결과를 볼 수 있었다.
이 논문에서 제공하는 데이터 분석의 소스 코드와 데이터 소스 링크가 전부 공개되어있어 시간을 두고 찬찬히 실습해보면서 어떻게 활용할지를 다시 고민해보려고 한다.
일단 지금 당장은, 이 논문에서 사용한 PANGO Lineage에 대해서 더 조사해보고 NCBI에서 찾을 수 있는 코로나 바이러스 염기 서열 데이터를 미리 쉽게 사용이 가능한 형태로 전처리하기로 했다. raw data는 염기 서열마다 유실된 부분이 있어 서열의 길이가 일정하지 않고 이 때문에 데이터를 바로 활용하기에 noise가 너무 많다는 문제점이 있을 것으로 예상되었다.
이에 Alignment를 실행하여 데이터 전처리를 하기로 했고 이 부분은 다른 팀원들이 담당해주기로 하였다.
나는 일단 Alignment가 실행된 이후의 일을 고민해보려고 했다. 우선은 NCBI에서 염기 서열이나 단백질 서열을 받아올 때 사용하는 FASTA라는 파일 형태를 다루기 위한 방법들을 찾아보았다.
1. R을 이용한 방법
짧고 빈 부분이 거의 없는 데이터인 뎅기열 바이러스의 염기 서열 데이터를 NCBI에서 받아 R의 Bioinformatics library인 SeqinR을 이용해 다음과 같은 간단한 전처리 과정을 실습했다.
https://www.ncbi.nlm.nih.gov/
-> 위 링크는 NCBI 링크이다. 여기서 뎅기열 바이러스를 검색창에 넣고 왼쪽 드롭다운 박스에서 Nucleotide로 설정한 다음 검색 결과 중 한 데이터를 받아서 사용했다.
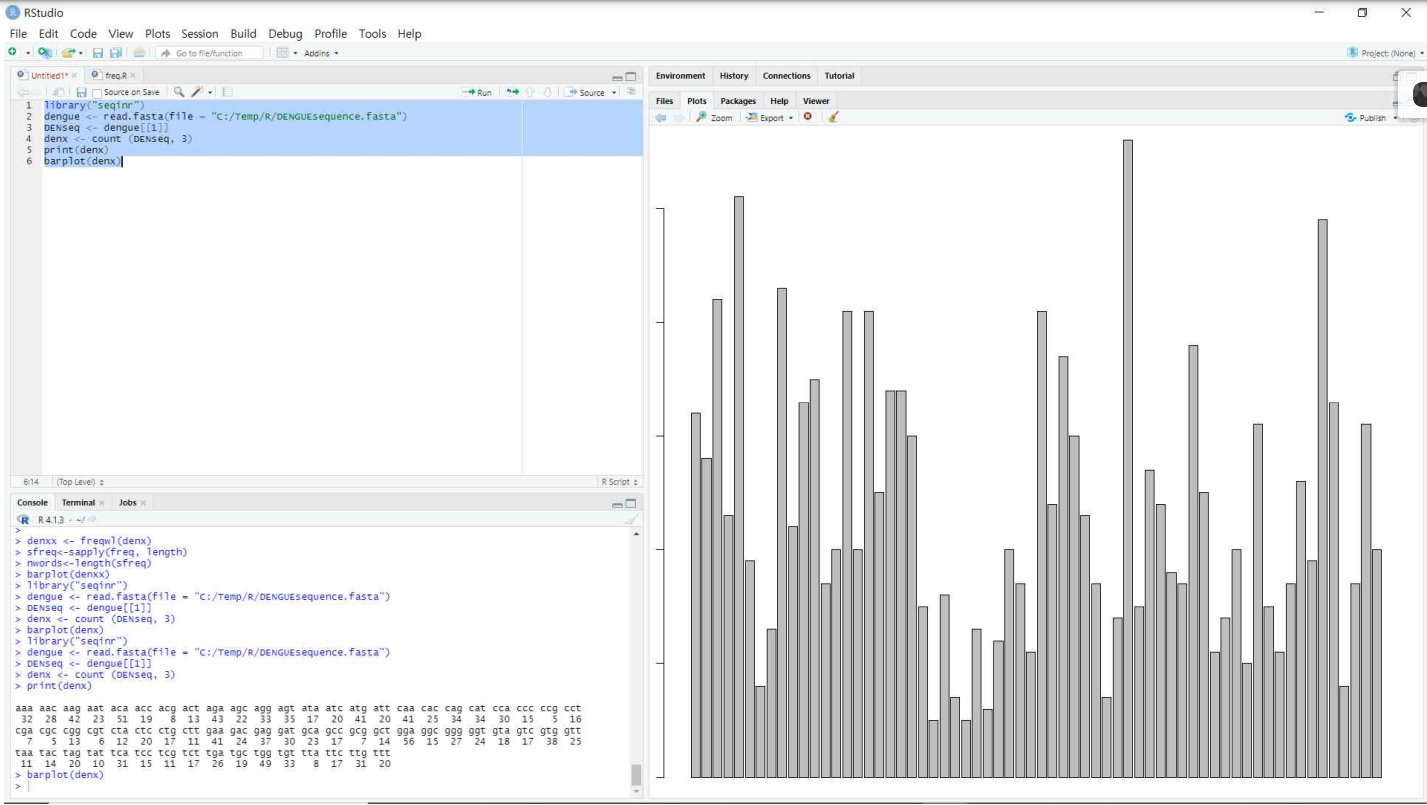
library("seqinr")
dengue <- read.fasta(file = "C:/Temp/R/DENGUEsequence.fasta")
DENseq <- dengue[[1]]
table(DENseq)
x <- count (DENseq, 3)
par("mar")
par(mar = c(1, 1, 1, 1))
plot(x)위와 같은 코드를 이용해 바이러스의 RNA 염기 서열을 전처리하면 이렇게 나온다. 코돈(RNA 염기 서열에서 하나의 아미노산을 표현하는 가장 작은 기본 단위)로 전체 서열을 분석하고 각 코돈의 빈도수를 표현한 결과가 차트로 나온다.
이외에도 SeqinR을 이용해서 할 수 있는 염기 서열 분석 방법을 찾아보려고 시도했으나 R을 이용하는 것은 방대한 염기 서열 데이터를 한 번에 처리하여 다루기에 한계가 있는 듯 했다. 그래서 다른 방법을 찾기 위해 도서를 찾던 중 다음의 책을 찾게 되었다.
바이오파이썬으로 만나는 생물정보학
2. BioPython을 이용한 분석 방법
파이썬 라이브러리 중에 BioPython이라는 것이 있고 pandas처럼 pip3로 install한 뒤에 import하여 사용할 수 있는 생명정보학을 위한 데이터 처리 함수들이 그 안에 내장되어있다고 한다.
로컬이 아니라 구글 드라이브에 대용량 파일을 넣어두고 런타임을 GPU로 설정하여 colab을 통한 빅데이터 처리도 가능하기 때문에 이 방법을 사용하는 것이 더 좋을 것이라 판단했다.
덤으로 염기 서열 문자열 데이터를 직접 연산하는 것은 상당히 오랜 시간이 걸리기 때문에 하나의 염기 서열 데이터를 허프만 코드를 이용해 이진코드로 압축한 다음 사용하는 것을 고려하고 있다.
이에 임시로 83개 문자열 길이의 염기 서열 FASTA파일을 다운받아 허프만 인코딩과 디코딩을 시도하는 실습을 다음과 같이 진행하였다.
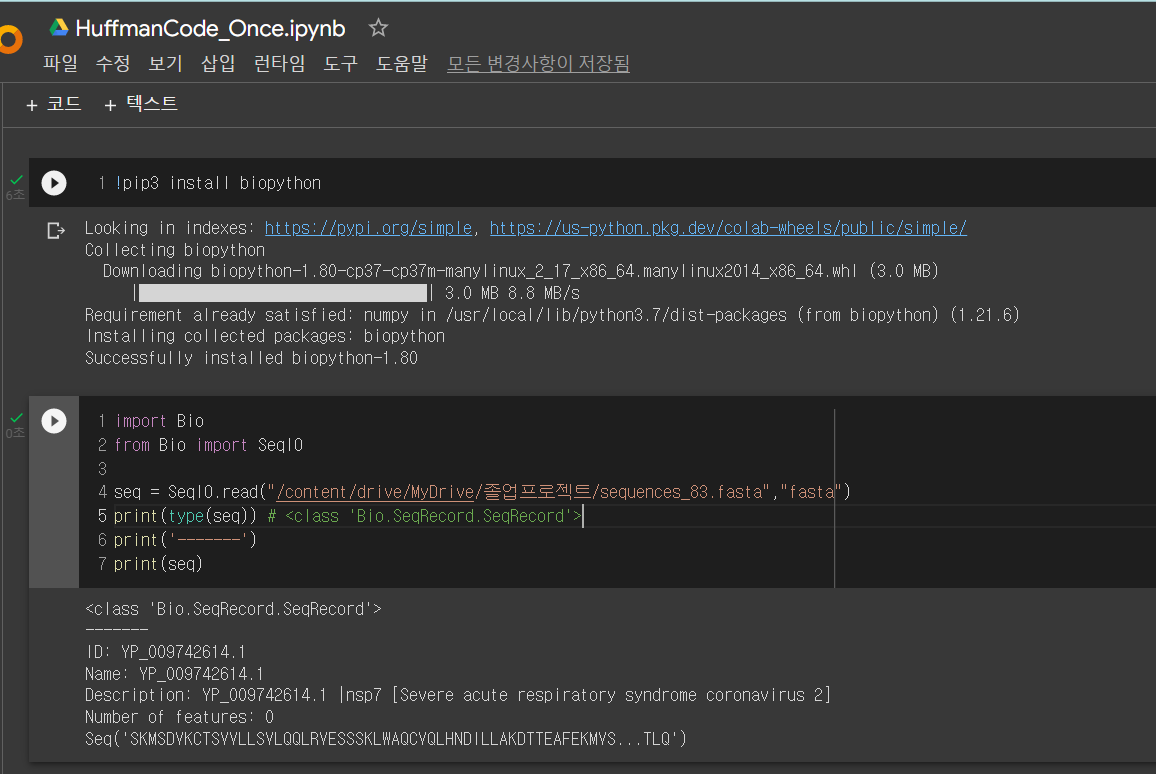
우선 biopython을 colab 해당 파일에 install 해주고 biopython에 내장된 함수를 이용해서 fasta 파일을 읽어온 것이다.
데이터의 ID, Name, Description, Number of features, Sequence가 출력되고 여기서 필요한 것은 Seq이다. 나머지는 메타 데이터이고 메타 데이터에 대해서는 다른 팀원들이 전처리를 통해 다운로드 받아 사용할 Sequence 데이터의 범위를 한정해줄 때 사용하게 될 것이다.
import heapq
from heapq import heappop, heappush
def isLeaf(root):
return root.left is None and root.right is None
# A 트리 노드
class Node:
def __init__(self, ch, freq, left=None, right=None):
self.ch = ch
self.freq = freq
self.left = left
self.right = right
# `__lt__()` 함수를 재정의하여 `Node` 클래스가 우선 순위 대기열과 함께 작동하도록 합니다.
# 가장 높은 우선순위 항목이 가장 낮은 빈도를 갖도록
def __lt__(self, other):
return self.freq < other.freq
# 허프만 트리를 탐색하고 사전에 허프만 코드를 저장
def encode(root, s, huffman_code):
if root is None:
return
#가 리프 노드를 찾았습니다.
if isLeaf(root):
huffman_code[root.ch] = s if len(s) > 0 else '1'
encode(root.left, s + '0', huffman_code)
encode(root.right, s + '1', huffman_code)
# 허프만 트리를 탐색하고 인코딩된 문자열을 디코딩합니다.
def decode(root, index, s):
if root is None:
return index
#가 리프 노드를 찾았습니다.
if isLeaf(root):
print(root.ch, end='')
return index
index = index + 1
root = root.left if s[index] == '0' else root.right
return decode(root, index, s)
#는 허프만 트리를 구축하고 주어진 입력 텍스트를 디코딩합니다
def buildHuffmanTree(text):
# 기본 케이스: 빈 문자열
if len(text) == 0:
return
#는 각 캐릭터의 출현 빈도를 계산합니다.
# 사전에 저장
freq = {i: text.count(i) for i in set(text)}
# Huffman 트리의 라이브 노드를 저장할 우선 순위 대기열를 만듭니다.
pq = [Node(k, v) for k, v in freq.items()]
heapq.heapify(pq)
# Queue에 노드가 둘 이상 있을 때까지 수행
while len(pq) != 1:
# 우선 순위가 가장 높은 두 노드를 제거합니다.
# Queue에서 #(최저 주파수)
left = heappop(pq)
right = heappop(pq)
#는 이 두 노드를 자식으로 사용하여 새 내부 노드를 만들고
#는 두 노드의 주파수 합과 동일한 주파수를 사용합니다.
# 우선 순위 대기열에 새 노드를 추가합니다.
total = left.freq + right.freq
heappush(pq, Node(None, total, left, right))
# `root` 는 허프만 트리의 루트에 대한 포인터를 저장합니다.
root = pq[0]
#는 Huffman 트리를 탐색하고 Huffman 코드를 사전에 저장합니다.
huffmanCode = {}
encode(root, '', huffmanCode)
# 허프만 코드 인쇄
print('Huffman Codes are:', huffmanCode)
print('The original string is:', text)
# 인코딩된 문자열을 인쇄합니다.
s = ''
for c in text:
s += huffmanCode.get(c)
print('The encoded string is:', s)
print('The decoded string is:', end=' ')
if isLeaf(root):
# 특수 케이스: a, aa, aaa 등과 같은 입력용
while root.freq > 0:
print(root.ch, end='')
root.freq = root.freq - 1
else:
#는 이번에도 허프만 트리를 횡단하고,
# 인코딩된 문자열 디코딩
index = -1
while index < len(s) - 1:
index = decode(root, index, s)
# Python에서 허프만 코딩 알고리즘 구현
if __name__ == '__main__':
buildHuffmanTree(seq)
print(len(seq))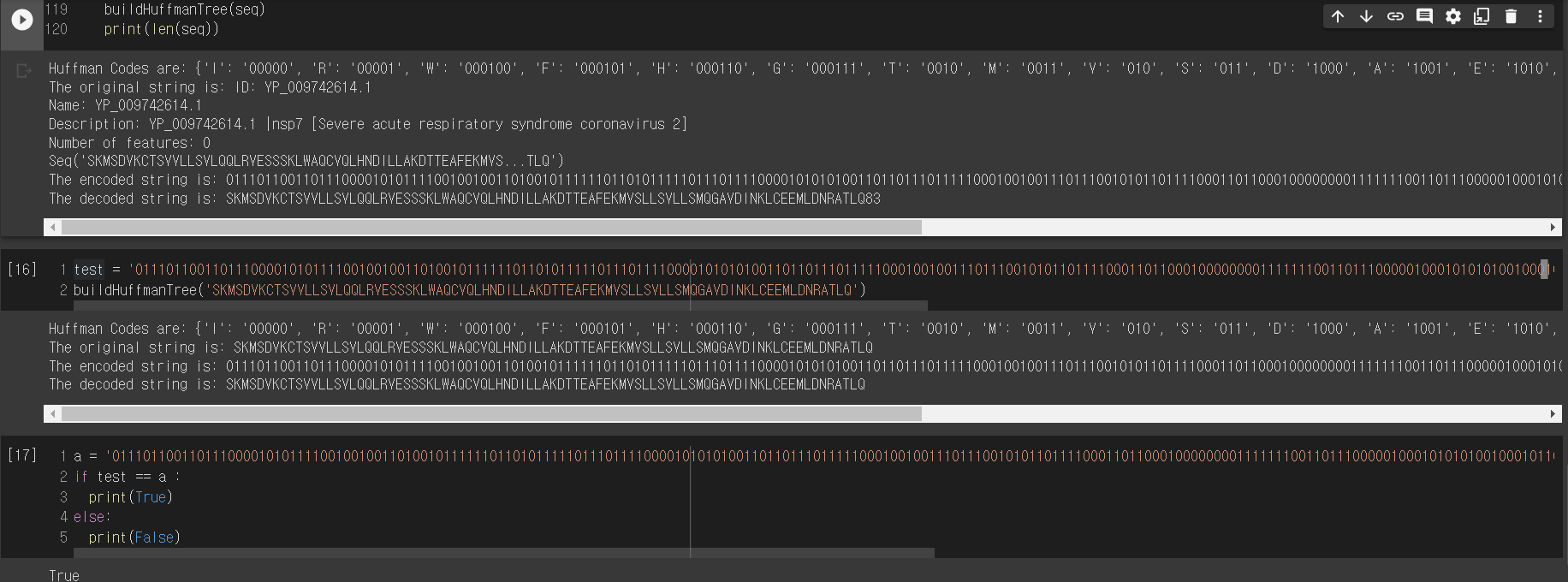
위의 허프만 코드를 이용해서 Sequence 데이터를 encoding, decoding한 후 혹시나 유실된 부분이나 오류가 없는지 다시 확인하기 위해서 디버깅 작업을 해본 것이다. 깔끔하게 encoding, decoding 과정이 진행되는 것을 확인했고 BioPython을 이용해서 할 수 있는 것을 잘 설명한 책이 중앙도서관에 비치되어있는 것을 확인하여 이에 관련된 실습을 진행하면서 앞으로 어떻게 데이터를 분석해 전처리하여 프로젝트에 반영할지 고민할 것이다.
** 일단은... PANGO Lineage에 대해서 좀 더 자세히 알고 나면 이를 적용해서도 데이터 처리를 해볼 수 있을 것 같다. 지금은 웹 사이트에서 새로운 염기 서열 데이터를 입력했을 때 기존 데이터들과의 비교와 분석 결과를 출력해줌으로써 기본적인 Bioinformatics 분석 결과를 제공해주는 것으로 프로젝트 방향을 구상해보고 있는 중이다.
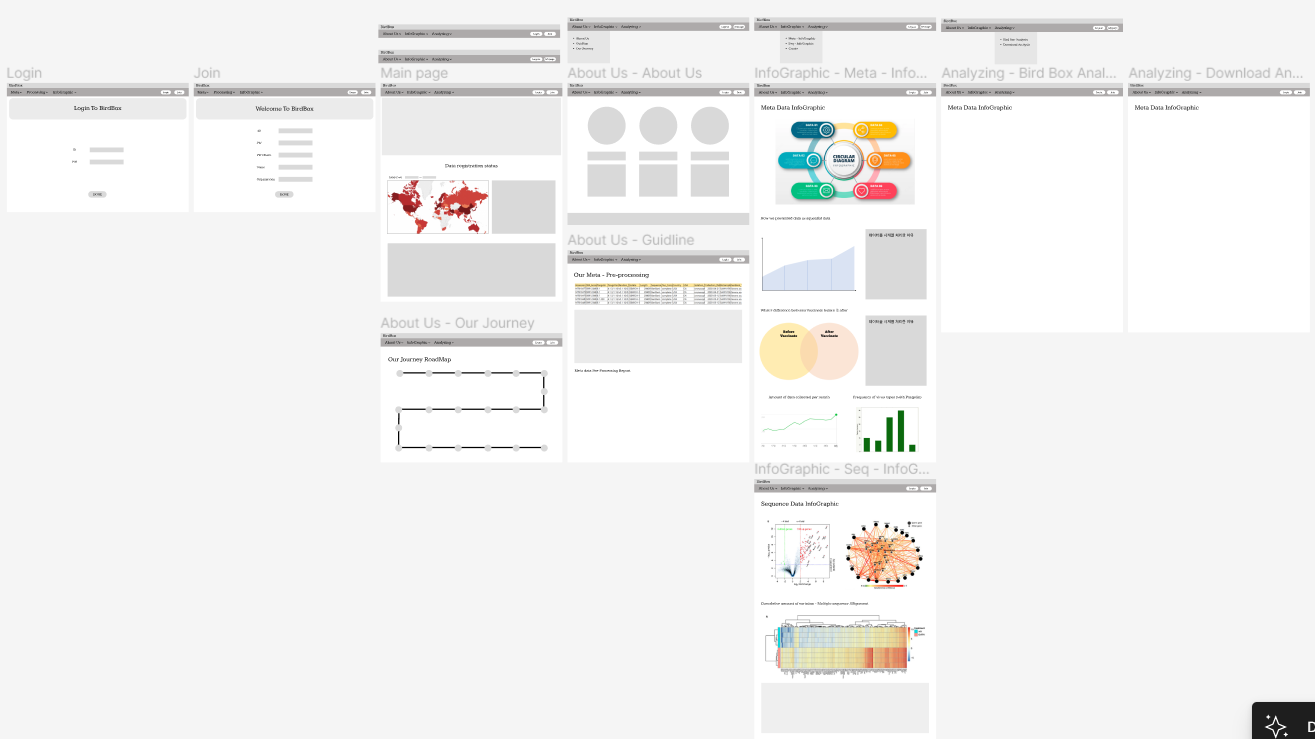
이것저것 자료들을 찾아보면서 제공할 서비스의 방향과 프로젝트 진행의 방향성을 담은 UI/UX를 간단하게 설계해두었다. 팀에서 분석한 내용들을 인포그래픽으로 제공하면서, 현재까지의 바이러스 분석 결과를 제공하고 이를 레포트 방식으로 출력하거나 새로운 데이터를 입력했을 때 그에 대한 분석 결과를 제공할 수 있는 사이트이다. multiple sequence alignment를 진행한 뒤에 누적된 변이의 양상을 시계열 데이터로 처리해서 이를 바탕으로 그 다음을 예측할 수 있는 그래프를 그리는 것이 목표가 되었다.
데이터 시각화 간단하게 해서 데이터 분석을 시도해봤다.
import pandas as pd
import matplotlib.pyplot as plt
csv_meta = pd.read_csv("/content/drive/MyDrive/2022-코딩 과제들/sequences.csv")
csv_meta.head()이렇게 코드를 쳐서 파일 형태를 확인해봤다.
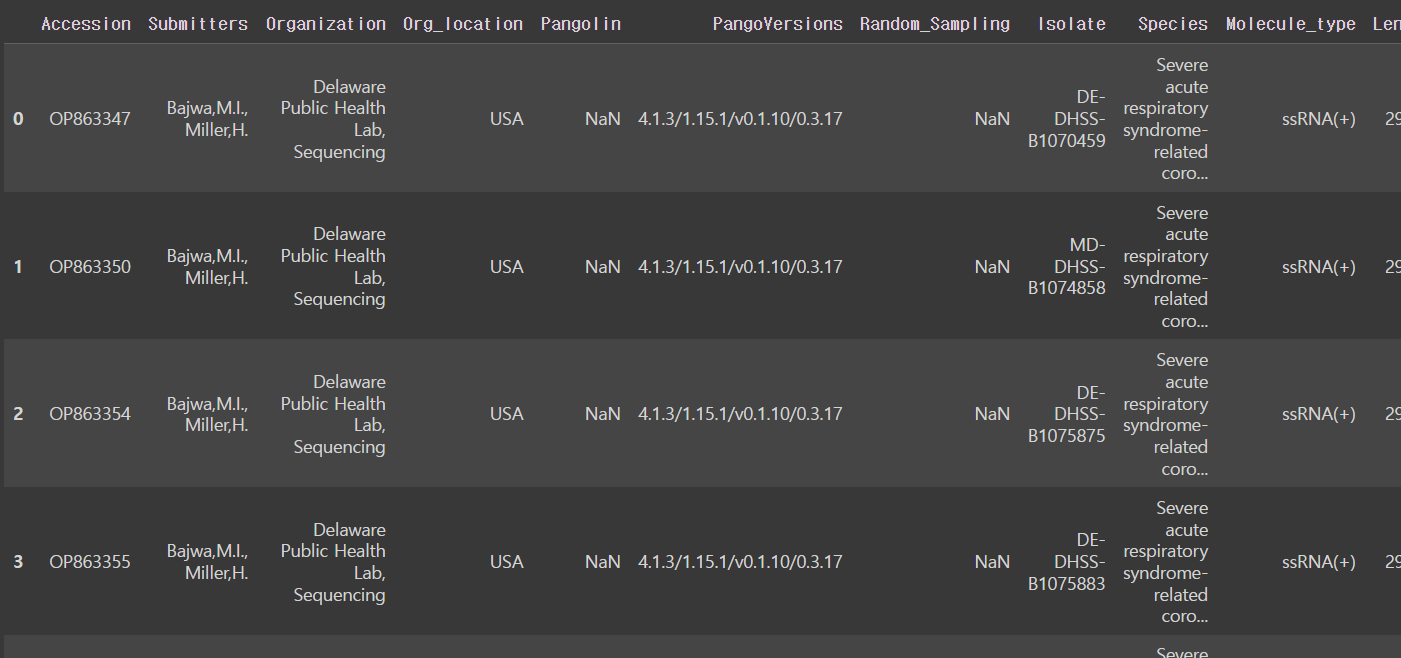
데이터가 대충 이렇게 생긴 것을 확인했으니 이 중에서 시각화를 위해서 보려고 하는 column만 선별해서 뽑아왔다.
csv_meta['Pangolin'].head()
Pangolin_data = pd.DataFrame(csv_meta.groupby('Pangolin').size())
Pangolin_data.head()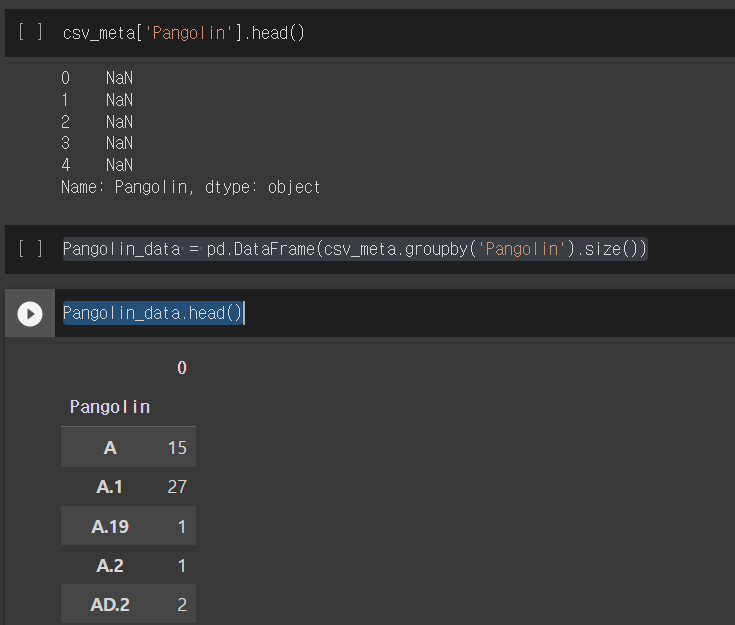
PANGO Lineage는 바이러스들의 변이를 분석해서 계통 분류를 해놓고 새로운 데이터에 대해서 그 데이터가 어떤 변이에 해당하는지를 분류하는 classifier AI이다. 분류 결과가 column에 저장되어있는데 따로 뜯어서 보니 NaN 값들이 있다. 아직 분류가 되지 않은 변이들인 것으로 추정된다.
Pangolin_data.columns = ['sum']
Pangolin_data.plot.bar(y='sum', rot=0)
ax = Pangolin_data.plot(kind='bar', title='Pangolin Frequency', figsize = (60, 4), legend = True, fontsize = 12)
column 명이 빈 것을 sum이라는 이름으로 채워주고 이를 plot 했는데... 데이터가 너무 많아서 값들이 겹치길래 이를 옆으로 넓게 좀 펼쳐주었다.
어떤 변이가 데이터셋 안에 얼마나 있는지를 확인하는 시각과 코드인데 이걸 다른 기간에 대한 데이터셋을 가지고도 실행시켜봤다.
대략적인 시계열적 특성이 있는 것 같아 이를 활용해보는 것으로 프로젝트 구상을 조정 중이다.

잘 읽었어요~