사실 리뷰가 아니라 모르는 개념들 정리에 가깝습니다.
기초가 있으시면 해당 링크를 참고하시면 됩니다.
참고
논문의 Main 파트에서 각 figure가 나오는 부분 전까지 핵심적인 내용을 요약하면 다음과 같습니다.
1. 서론
AlphaFold는 단백질의 3차원 구조를 예측하는 데 있어서 기존 방법들보다 월등한 성과를 보여주는 모델입니다. 단백질 구조는 그 기능을 이해하는 데 매우 중요한데, 현재까지 약 100,000개의 단백질 구조가 실험적으로 밝혀졌으나 이는 전체 단백질 서열 중 극히 일부분에 해당합니다. AlphaFold는 계산적 접근을 통해 이러한 문제를 해결하려는 목적으로 개발되었으며, 특히 유사한 구조가 알려지지 않은 단백질의 원자 수준의 정확도까지 예측할 수 있습니다.
2. 두 가지 단백질 구조 예측 접근법
단백질 구조 예측에는 크게 두 가지 접근 방식이 존재합니다:
1. 물리적 상호작용 기반 접근: 단백질의 분자 동력학 및 열역학적 상호작용을 시뮬레이션하여 구조를 예측합니다. 그러나 계산 복잡성 및 정확한 물리 모델의 부족으로 인해 이 방법은 큰 단백질에 대해서는 현실적이지 않습니다.
2. 진화적 역사 기반 접근: 단백질의 진화적 상호작용과 상동 구조를 기반으로 예측하는 방법입니다. 최근 유전체 데이터의 폭발적 증가와 딥러닝 기술 덕분에 이 방법의 성능이 크게 향상되었으나, 여전히 실험적 정확도에는 미치지 못하는 경우가 많습니다.
3. AlphaFold의 접근 방식
AlphaFold는 단백질 구조 예측을 위해 기존 방법의 한계를 극복하는 새로운 기계 학습 모델을 제시합니다. 이 모델은 다중 서열 정렬(MSA)과 단백질 구조의 물리적 및 생물학적 제약을 결합한 새로운 신경망 구조를 활용합니다. 이를 통해 AlphaFold는 실험적으로 규명된 구조와 비교할 수 있을 정도로 높은 정확도를 제공합니다.
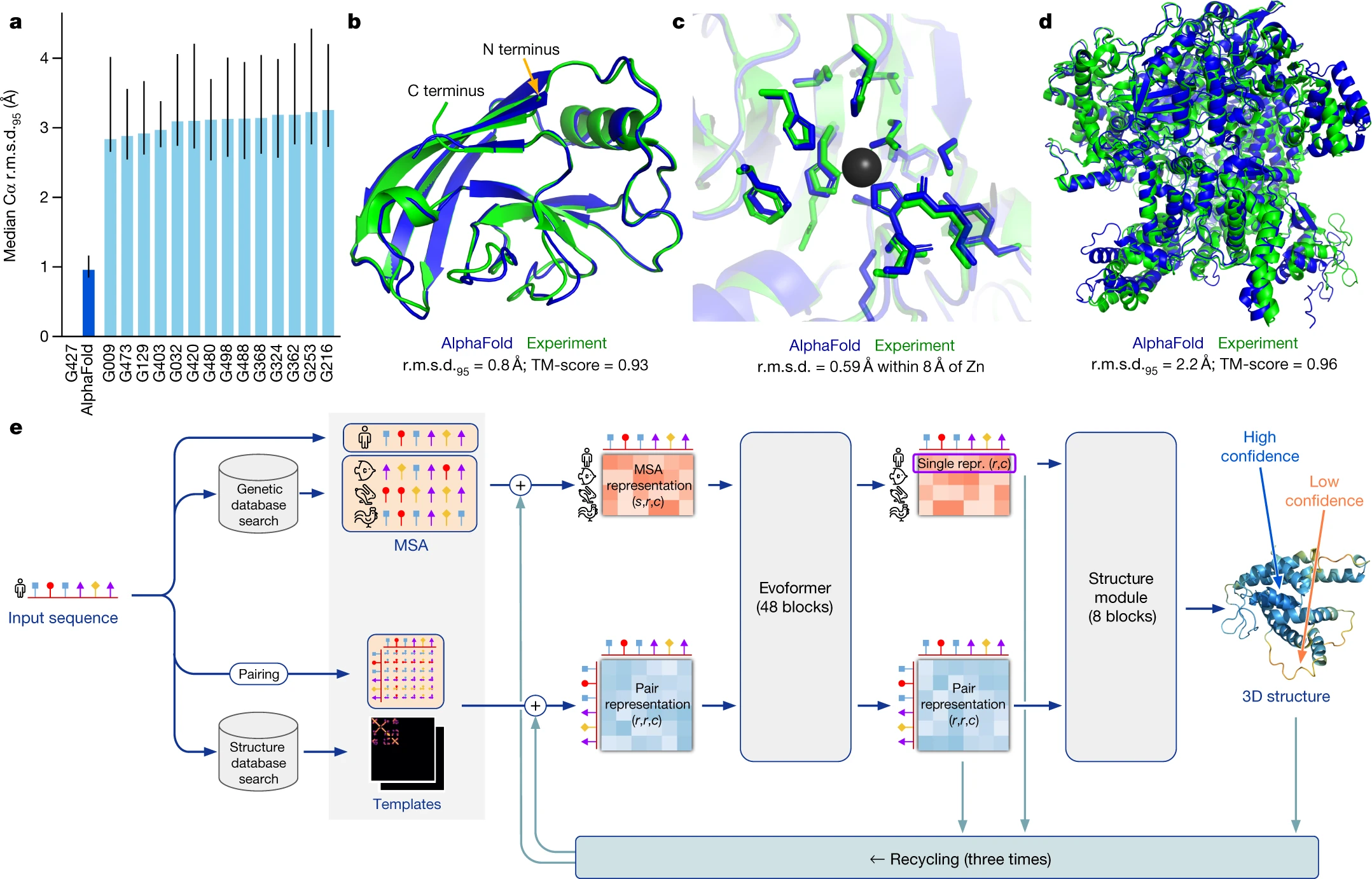
알파폴드가 예측한 단백질 구조와 실제 단백질 구조의 비교 이미지
4. CASP14에서의 성능
AlphaFold는 2020년 5월부터 7월까지 진행된 CASP14 대회에서 기존의 다른 모든 방법을 능가하는 성과를 보였습니다. AlphaFold의 단백질 구조 예측 정확도는 평균적으로 다른 방법들보다 크게 우수했으며, 특히 백본 구조와 측쇄의 예측에서 뛰어난 성능을 입증했습니다.
평가기준이 처음 들어보는 단어들이라 물어보니 다음과 같다 합니다.
1. Root-Mean-Square Deviation (r.m.s.d.)
- r.m.s.d.는 두 구조 간의 평균적인 거리 차이를 측정하는 값입니다. 두 구조를 비교할 때, 예측된 구조와 실제 구조 사이에서 각 원자(또는 알파 탄소) 위치의 차이를 계산합니다.
- 공식은 다음과 같습니다:여기서 은 비교하고 있는 원자의 수,는 각 원자쌍(예측된 위치와 실제 위치) 사이의 거리입니다.
- 즉, 두 구조의 원자들이 얼마나 가까이 위치해 있는지 또는 얼마나 일치하는지를 측정하는데, r.m.s.d. 값이 작을수록 예측된 구조가 실제 구조와 더 가깝습니다. 예를 들어, 1.46Å이라는 값은 두 구조의 평균적인 원자 간 거리가 1.46 옹스트롬(Å)임을 나타냅니다.
2. 알파 탄소 원자 (Cα)
- 알파 탄소(Cα)는 단백질의 기본 구조에서 각 아미노산 잔기의 중심에 위치한 원자입니다. 아미노산의 알파 탄소는 아미노산의 모든 부분(측쇄, 카복실기, 아미노기 등)을 연결하는 중요한 결합점입니다.
- 단백질의 구조는 주로 알파 탄소들의 위치에 따라 그 기본적인 형상을 이루기 때문에, 단백질 구조 예측에서 알파 탄소의 위치를 정확하게 예측하는 것이 중요합니다.
3. 옹스트롬 (Å)
- 옹스트롬(Å)는 원자와 분자 수준에서 길이를 측정하는 단위로, 1Å는 0.1 나노미터(10^-10 미터)에 해당합니다. 원자 간의 결합 길이나 분자의 크기 등을 나타낼 때 자주 사용됩니다. 예를 들어, 1.46Å는 원자 간의 거리가 평균적으로 1.46 옹스트롬임을 의미합니다. 이는 대략 두 원자 사이의 거리 정도로, 매우 작은 단위입니다.
4. pLDDT와 lDDT
- pLDDT (Predicted Local Distance Difference Test): AlphaFold가 구조 예측을 할 때 각 잔기에 대해 예측된 구조의 신뢰도를 나타내는 점수입니다. 이 값은 AlphaFold가 예측한 구조가 얼마나 정확할 것인지를 예측하는 척도입니다. pLDDT 점수가 높을수록 예측의 신뢰도가 높다고 판단할 수 있습니다. 보통 0에서 100 사이의 값을 가지며, 90 이상이면 매우 높은 신뢰도를 뜻합니다.
- lDDT (Local Distance Difference Test): 실제 구조와 예측된 구조 간의 현지적 정확도를 측정하는 지표입니다. 각 잔기 사이의 거리가 얼마나 일치하는지 측정하며, 예측된 구조가 실제 구조와 어느 정도 가까운지를 정량적으로 평가합니다. lDDT-Cα는 알파 탄소들 간의 거리에 기반한 lDDT 측정 방식입니다. AlphaFold는 pLDDT를 이용해 예측된 신뢰도를 제공하고, lDDT를 통해 그 정확도를 검증합니다.
5. pLDDT와 lDDT의 차이
- pLDDT는 AlphaFold가 자체적으로 예측한 신뢰도이며, lDDT는 실제 실험 데이터와 비교했을 때의 객관적인 정확도를 나타냅니다. 둘이 강한 상관관계를 가지는 것이 이상적이며, AlphaFold는 이를 잘 충족시키고 있습니다.
요약:
- r.m.s.d.는 예측된 구조와 실제 구조 사이의 평균적인 거리 차이를 나타내는 지표로, 값이 낮을수록 더 정확한 예측을 의미합니다.
- 알파 탄소(Cα)는 단백질 구조에서 각 아미노산의 중심이 되는 원자입니다.
- Å(옹스트롬)는 원자 수준의 거리를 나타내는 단위로, 매우 작은 길이를 측정합니다.
- pLDDT는 예측된 구조의 신뢰도를, lDDT는 실제 구조와의 비교를 통해 예측의 정확도를 나타냅니다.
5. AlphaFold의 신경망 구조
AlphaFold의 핵심 요소는 새로운 신경망 아키텍처인 Evoformer입니다. 이 네트워크는 MSA 데이터를 효과적으로 처리하여 단백질 서열과 잔기 사이의 상호작용을 예측합니다. Evoformer 블록은 서열과 잔기 쌍의 특성을 결합하여 단백질의 3차원 구조를 연속적으로 개선해 나가는 과정을 거칩니다. 또한 구조 모듈이 포함되어 있어 잔기의 회전 및 번역을 모델링하고, 이를 통해 더 정밀한 3D 구조를 생성합니다.
AlphaFold는 이 재귀적 학습 방식을 통해 반복적으로 예측을 개선하며, 궁극적으로는 실험적 구조와 거의 동일한 수준의 정확한 예측을 달성합니다.
6. 측쇄 예측 및 신뢰도 평가
AlphaFold는 단백질의 백본뿐만 아니라 측쇄의 위치도 정확하게 예측하며, 각 잔기의 신뢰도를 평가할 수 있는 기능도 제공합니다. 이를 통해 예측 결과의 정확성에 대해 신뢰할 수 있는 판단을 내릴 수 있습니다.
계속해서 AlphaFold 논문의 핵심적인 내용을 이어 설명하겠습니다.
7. CASP14 결과의 확장성
AlphaFold는 CASP14에서 뛰어난 성능을 보였을 뿐만 아니라, CASP14에서 훈련된 모델이 새로운 PDB 구조에도 높은 정확도를 유지하는지 검증했습니다. AlphaFold는 훈련 데이터에 포함되지 않은 새로운 PDB 구조들을 테스트했을 때도 예측 정확도가 매우 높았으며, 특히 백본 및 측쇄 구조의 정확도가 모두 유지되었습니다. 또한, AlphaFold는 잔기별로 예측의 신뢰도를 제공하여, 예측된 구조의 신뢰성을 평가할 수 있게 합니다.
8. AlphaFold의 신경망 아키텍처
AlphaFold의 예측 정확도를 크게 향상시킨 요소 중 하나는 신경망의 새로운 아키텍처와 훈련 방식입니다. AlphaFold는 단백질 구조의 진화적, 물리적, 기하학적 제약을 신경망에 반영하여, 더 정밀한 3D 구조 예측을 가능하게 합니다. 이 아키텍처의 중요한 특징 중 하나는 다중 서열 정렬(MSA)과 잔기 간 상호작용을 공동으로 임베딩하는 방식입니다. MSA는 진화적으로 관련된 서열들을 활용해 단백질 구조를 더 정확하게 예측하는 데 기여합니다.
AlphaFold의 구조 모듈은 잔기의 회전과 번역을 예측하여 3차원 공간에서 단백질의 구체적인 위치를 결정합니다. 이 모듈은 초기 상태에서는 모든 잔기를 동일한 위치에서 시작하지만, 반복적인 학습을 통해 점진적으로 정확한 구조를 형성해 나갑니다. AlphaFold는 재귀적 학습(recycling)을 통해 여러 번의 반복 후 구조 예측을 개선합니다.
AlphaFold 네트워크는 심층 학습을 기반으로 한 모델로, 단백질의 3차원 구조를 매우 정확하게 예측할 수 있도록 설계되었습니다. 여기서는 네트워크의 핵심적인 수학적 구성요소와 다중 서열 정렬(MSA: Multiple Sequence Alignment)에 대해 더 자세히 설명하겠습니다.
1. AlphaFold의 수학적 구성 요소
AlphaFold는 여러 가지 신경망 블록을 결합하여 단백질 서열의 진화적, 물리적, 기하학적 정보를 통합해 3D 구조를 예측합니다. 주요 구성 요소는 다음과 같습니다:
a. Evoformer 블록
Evoformer는 MSA(다중 서열 정렬)와 잔기 쌍 간(pairwise) 상호작용을 처리하는 블록으로, 단백질 구조 예측에 핵심적인 역할을 합니다. Evoformer는 두 가지 입력 데이터, 즉 MSA 데이터와 잔기 간 상호작용 데이터를 통합하여 단백질의 3차원 구조를 예측합니다.
-
MSA 입력 처리:
- 는 MSA에 포함된 서열의 수, 는 단백질의 잔기(residue) 수, 는 채널 수입니다.
- MSA는 각 서열에서 특정 위치에 있는 아미노산 정보를 행렬로 표현하며, 이는 잔기 간 진화적 연관성을 반영합니다.
-
Pairwise 입력 처리:
- 잔기 간 상호작용 데이터를 나타내는 행렬입니다. 이 행렬은 개의 잔기들 사이의 상호작용을 설명하며, 기하학적 거리나 물리적 상호작용을 반영할 수 있습니다.
-
Evoformer 계산 과정:
- Evoformer는 MSA 표현과 pairwise 표현을 교환하며 정보 업데이트를 진행합니다. MSA에서 진화적 상관관계와 정보를 추출하고, 그 결과를 잔기 간 상호작용을 처리하는 데 사용합니다.
Evoformer 블록은 Axial Attention이라는 메커니즘을 사용합니다. Axial Attention은 MSA 및 잔기 쌍의 정보를 각각 독립적으로 처리한 후, 두 정보가 결합되도록 구성되어 있습니다.
- MSA에서 잔기 간 상호작용에 대한 정보를 반영할 때, 행렬 곱으로 각 잔기들의 정보가 상호작용합니다. 이 때 MSA의 각 행에 있는 서열 간 상호작용이 잔기 간 상호작용을 갱신하는 데 사용됩니다.
여기서 는 -번째 Evoformer 블록에서의 MSA 상태이고, 는 잔기 간 상호작용 상태입니다.
b. Invariant Point Attention (IPA)
IPA는 단백질의 3차원 좌표를 직접 예측하기 위해 개발된 메커니즘입니다. 이 블록은 단백질의 잔기들 사이의 기하학적 위치 관계를 정확하게 모델링합니다. IPA는 3차원 공간에서 각 잔기의 위치를 다음과 같은 방식으로 추정합니다.
- 입력: 각 잔기의 위치는 회전 및 번역으로 표현됩니다. 잔기 의 위치는 (3차원 회전 행렬)과 (3차원 번역 벡터)로 표현됩니다.
- Attention 기법: IPA는 attention 메커니즘을 통해 각 잔기의 회전과 번역을 예측합니다. Attention은 전통적인 query, key, value 방식을 사용하지만, 잔기 간 거리와 회전을 기반으로 하는 변형된 attention을 사용합니다.
- 결과: 잔기 의 새로운 위치는 다음과 같이 계산됩니다.여기서 , , 는 각 잔기에 대한 query, key, value입니다.
c. 재귀적 학습 (Recycling)
AlphaFold는 구조 예측을 한 번에 끝내지 않고, 재귀적 학습을 통해 예측 결과를 반복적으로 개선합니다. 재귀적 학습 과정에서, 이전에 예측된 구조를 다시 네트워크에 입력으로 제공하여, 점진적으로 더 정확한 예측을 할 수 있도록 합니다. 이 과정은 기계 학습에서 일반적인 "iterative refinement" 방식과 유사합니다.
2. MSA (Multiple Sequence Alignment, 다중 서열 정렬)
다중 서열 정렬(MSA)은 여러 단백질 서열을 정렬하여, 이들 사이의 진화적 상관관계를 분석하는 방법입니다. MSA는 AlphaFold에서 매우 중요한 입력으로, 서열 간의 상동성을 바탕으로 단백질 구조를 예측하는 데 큰 도움을 줍니다.
a. MSA 구성
MSA는 단백질 서열을 다음과 같이 배열합니다:
- 각 열(column): 특정 위치에 있는 아미노산을 나타냅니다. 이 열은 해당 위치에서 진화적 보존성을 보여줍니다.
- 각 행(row): 서로 다른 단백질 서열을 나타냅니다. 이 서열들은 진화적 유사성을 가지며, 상동성(homology)을 바탕으로 정렬됩니다.
b. MSA의 역할
MSA는 특정 단백질 서열이 진화 과정에서 어떻게 변화해왔는지를 보여줍니다. 중요한 아미노산들은 진화적으로 보존되기 때문에, MSA에서 동일한 위치에 나타날 가능성이 높습니다. 이를 통해 AlphaFold는 단백질의 구조적 제약을 더 잘 파악할 수 있습니다.
c. MSA에서의 상호작용 분석
MSA에서 특정 아미노산들이 진화적으로 자주 함께 나타나는 패턴은 잔기들 간의 상호작용을 암시합니다. AlphaFold는 이러한 상관관계를 학습하여, 잔기들 사이의 거리를 더 정확하게 예측할 수 있습니다. MSA 기반 정보는 잔기들 간의 공진화(co-evolution) 패턴을 통해 단백질의 3차원 구조를 유추하는 데 큰 역할을 합니다.
요약
AlphaFold 네트워크는 Evoformer 블록과 IPA를 통해 단백질 서열에서 직접 3차원 구조를 예측하며, MSA를 활용해 단백질 서열 간의 진화적 상관관계를 분석합니다. 수학적으로는 MSA와 잔기 간의 pairwise interaction 정보를 Axial Attention을 사용해 결합하고, IPA로 3D 구조를 예측합니다.

9. Evoformer의 역할
AlphaFold의 주요 신경망 블록인 Evoformer는 단백질 구조 예측의 중심적 역할을 합니다. Evoformer는 단백질의 3차원 구조를 그래프 추론 문제로 보고, 잔기들 간의 상호작용을 예측합니다. 잔기들 간의 쌍 관계(pair representation)를 업데이트하면서 단백질 구조가 점진적으로 형성됩니다.
Evoformer는 서열 기반(MSA)과 잔기 쌍 간(pair-wise)의 정보를 지속적으로 교환하면서 구조 예측을 수행합니다. Evoformer의 혁신적인 점은 이러한 정보 교환을 매 반복에서 수행함으로써 예측의 정밀도를 높인다는 것입니다. 이를 통해 AlphaFold는 단백질 서열과 잔기 간의 상호작용을 종합적으로 분석하여 3D 구조를 성공적으로 예측할 수 있습니다.
Evoformer는 AlphaFold의 핵심 블록으로, 단백질 서열(다중 서열 정렬, MSA)과 잔기 간 상호작용 정보를 처리하여 단백질의 3차원 구조를 예측하는 데 중요한 역할을 합니다. Evoformer는 Axial Attention과 다양한 업데이트 메커니즘을 통해 정보 교환을 수행하며, 특히 잔기 간 상호작용(pairwise interaction)과 서열 간 상호작용을 통합합니다.
1. Evoformer의 입력
Evoformer는 두 가지 주요 입력을 처리합니다:
1. MSA 행렬
-: 다중 서열 정렬(MSA)에 포함된 서열의 수
-: 단백질 잔기의 수
-: 각 서열과 잔기의 특징 벡터의 차원
2. Pairwise 행렬
-: 단백질 잔기의 수
-: 잔기 간 상호작용을 나타내는 특징 벡터의 차원
MSA 행렬은 각 잔기 위치에서 여러 단백질 서열을 정렬한 것이며, pairwise 행렬은 잔기들 간의 상호작용 정보를 표현합니다.
2. Axial Attention
Axial Attention은 Evoformer의 핵심 메커니즘 중 하나로, 잔기 간 상호작용과 MSA 간 상호작용을 처리하는 데 사용됩니다. 이는 전통적인 attention 메커니즘을 변형하여 두 가지 축(서열 축과 잔기 축)을 따로따로 처리합니다.
a. Axial Attention의 수학적 표현
Axial Attention은 두 축(서열 및 잔기)에 대해 각각의 attention을 수행합니다. attention 메커니즘의 일반적인 수식은 다음과 같습니다:
여기서:
-: query 행렬 (각 잔기에 대한 특징)
-: key 행렬 (각 잔기에 대한 특징)
-: value 행렬 (각 잔기에 대한 정보를 포함)
-: query와 key의 차원
Axial Attention은 이를 서열 및 잔기 차원에 대해 각각 독립적으로 수행합니다:
1. 서열 축(MSA 차원)에 대한 Attention:
여기서는 서열 정보를 나타내는 행렬입니다. MSA 행렬의 각 잔기 위치에 대해, 다른 서열들로부터 정보를 가져와 업데이트합니다.
- 잔기 축(Pairwise 차원)에 대한 Attention:여기서는 잔기 간 상호작용 정보를 나타내는 행렬입니다. 각 잔기 위치에 대해 다른 잔기들과의 상호작용 정보를 고려하여 업데이트합니다.
Axial Attention을 통해 각 잔기 간의 관계와 서열 간의 관계를 독립적으로 처리한 후, 이를 결합하여 최종적으로 잔기 간 상호작용과 서열 정보를 결합합니다.
3. MSA 및 Pairwise 업데이트
Evoformer는 MSA와 Pairwise 정보 간의 교환을 통해 단백질의 구조적 제약을 반영합니다.
a. Outer Product Mean
MSA에서 진화적 상호작용을 반영하기 위해, Evoformer는 각 잔기 위치에서 MSA 행렬의 행벡터 간 외적(outer product)을 계산합니다. 이 과정은 MSA 정보가 pairwise interaction에 영향을 미치도록 합니다.
외적 계산은 다음과 같이 표현될 수 있습니다:
여기서는 서열에서의 잔기 위치 벡터입니다.
이를 통해, MSA 행렬의 각 서열에 있는 잔기들이 pairwise 상호작용에 기여하는 방식을 모델링합니다.
b. Pairwise Representation 업데이트
잔기 간 상호작용을 나타내는 행렬은 Evoformer에서 여러 차례 반복적으로 업데이트됩니다. 이는 Axial Attention 외에도 다양한 메커니즘을 통해 처리됩니다.
삼각형 곱셈 업데이트(Triangle Multiplicative Update)와 삼각형 주의 삼각형(Triangle Attention)은 잔기 간 상호작용을 개선하는 데 사용됩니다. 이 과정은 잔기 간의 거리가 삼각형 부등식을 만족하는지 등의 제약을 반영하여, 더 정밀한 잔기 간 관계를 학습하게 합니다.
c. Layer Normalization 및 Feed-Forward 네트워크
각 단계에서의 업데이트 후, Layer Normalization과 Feed-Forward Network (FFN)이 적용됩니다. 이는 다음과 같이 표현됩니다:
여기서는 MSA 또는 pairwise 행렬이며, attention을 통한 업데이트 후, FFN을 통해 비선형 변환이 적용됩니다.
4. Evoformer의 전체 과정
- MSA 행렬과 Pairwise 행렬을 초기화합니다.
- Axial Attention을 통해 MSA와 잔기 간 상호작용을 처리합니다.
- Outer Product Mean을 계산하여, MSA의 정보가 pairwise 상호작용에 반영되도록 합니다.
- Triangle Multiplicative Update와 Triangle Attention을 통해 잔기 간의 관계를 개선합니다.
- Layer Normalization과 Feed-Forward Network를 통해 업데이트된 정보를 정규화하고, 비선형 변환을 적용하여 최종 구조를 예측합니다.
요약
Evoformer는 Axial Attention을 사용하여 MSA(다중 서열 정렬)와 pairwise 잔기 간 상호작용을 처리하며, 이를 통해 단백질의 3차원 구조를 예측하는 데 필요한 중요한 정보를 추출합니다. 외적(outer product)을 계산하여 서열 간 상호작용을 잔기 간 상호작용으로 변환하고, 삼각형 곱셈과 주의 메커니즘을 통해 잔기 간 거리 제약을 학습합니다. Evoformer의 이 과정들은 반복적으로 수행되어 점진적으로 더 정확한 구조 예측을 가능하게 합니다.
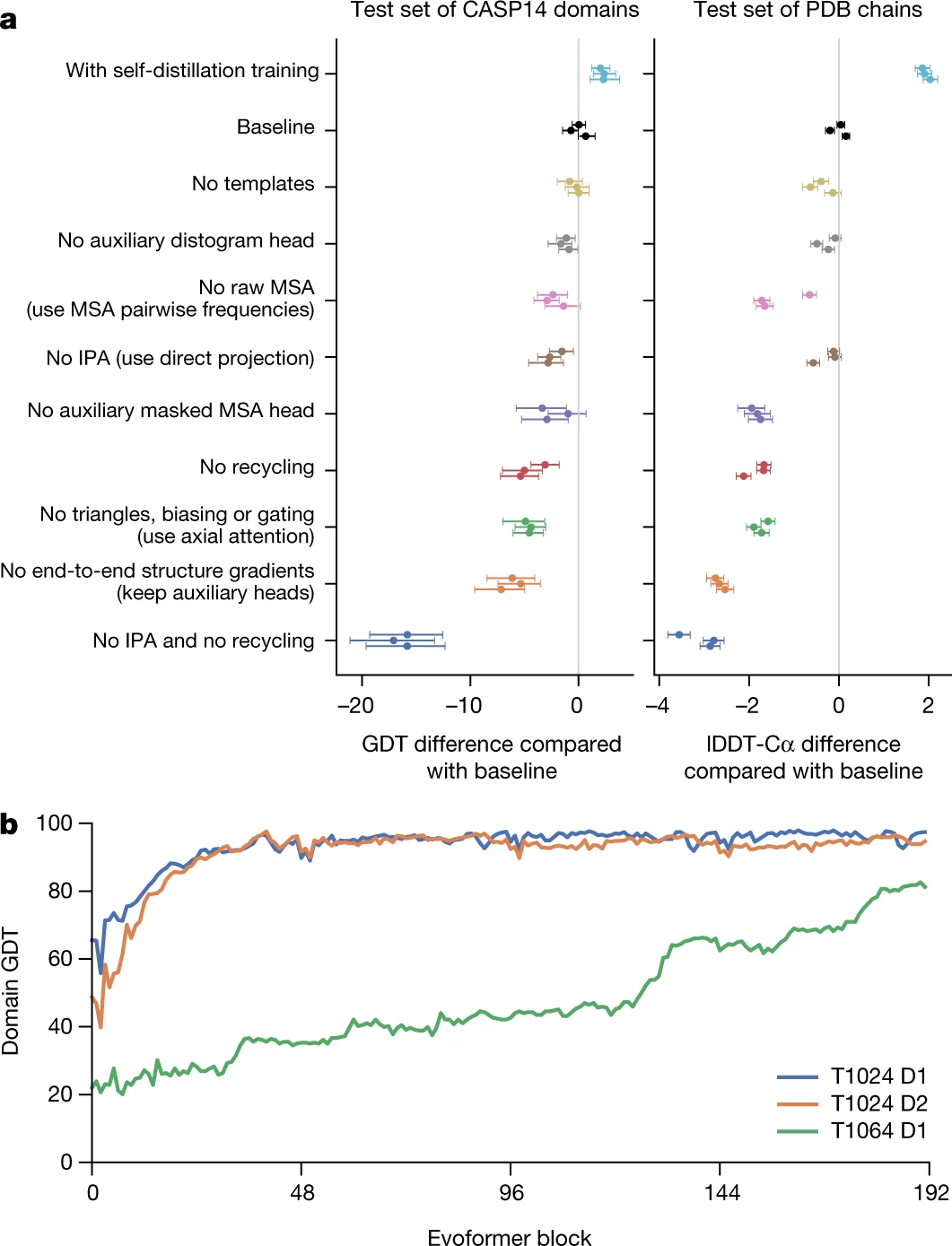
10. 구조 예측의 끝 단계
AlphaFold의 구조 모듈은 단백질의 백본과 측쇄를 동시에 예측하며, Invariant Point Attention (IPA) 모듈을 사용해 3D 공간에서의 각 잔기의 위치를 정교하게 추정합니다. IPA는 각 잔기의 회전과 번역을 예측하고, 이를 통해 단백질의 전체적인 구조를 완성합니다. 이 과정에서 AlphaFold는 다양한 회전 및 번역 패턴을 고려하여, 잔기들이 실제로 배치될 위치를 정밀하게 계산합니다.
11. 예측의 신뢰도와 추가적인 성능 향상
AlphaFold는 예측된 구조에 대해 pLDDT (predicted Local Distance Difference Test) 점수를 제공하며, 이 점수는 예측된 구조의 정확성을 평가하는 기준이 됩니다. 또한, TM-score는 예측된 구조와 실제 구조 간의 전역적 유사성을 측정하며, 이를 통해 AlphaFold의 예측이 얼마나 정확한지를 평가할 수 있습니다.
AlphaFold는 재귀적 학습과 구조 모듈을 통해 예측된 구조의 정확도를 지속적으로 개선하며, 특히 실험적으로 규명되지 않은 새로운 단백질 서열에 대해서도 높은 신뢰성을 제공합니다. 이러한 점에서 AlphaFold는 단백질 구조 예측의 중요한 돌파구로 평가받고 있습니다.
여기까지가 AlphaFold 모델의 핵심적인 내용입니다. 이후 논문은 AlphaFold의 구체적인 신경망 아키텍처와 구조적 세부 사항을 설명하며, 다양한 실험 결과와 함께 모델의 성능을 입증합니다.
계속해서 AlphaFold 논문의 다음 핵심적인 내용을 설명하겠습니다.
12. 훈련 방법 및 데이터 사용
AlphaFold는 대규모의 단백질 구조 데이터를 기반으로 훈련되었으며, 두 가지 주요 데이터 소스를 사용했습니다:
1. PDB 데이터(Protein Data Bank), 이는 실험적으로 밝혀진 단백질 구조의 데이터입니다.
2. Uniclust30 및 BFD와 같은 대규모 서열 데이터베이스, 이는 단백질 서열 간의 진화적 상호작용을 파악하는 데 사용되었습니다.
AlphaFold의 신경망은 PDB 데이터로만 훈련해도 높은 정확도를 제공할 수 있지만, self-distillation이라는 기법을 통해 더욱 높은 정확도를 달성할 수 있었습니다. Self-distillation은 이미 훈련된 모델을 사용해 새로운 단백질 서열을 예측한 후, 이 예측된 구조를 사용해 모델을 다시 훈련시키는 방식입니다. 이를 통해 훈련 데이터가 없는 경우에도 예측 정확도를 높일 수 있습니다.
또한, AlphaFold는 MSA 데이터에서 무작위로 일부 서열을 가리거나 돌연변이를 넣는 방식으로 추가적인 학습 목표를 설정해, 단백질 서열 간의 진화적 상관관계를 더 잘 해석할 수 있게 했습니다. 이를 통해 AlphaFold는 MSA 데이터를 더 효과적으로 활용하고, 더 정확한 구조를 예측할 수 있게 됩니다.
13. 신경망 해석 및 구조 예측 과정
AlphaFold가 단백질 구조를 어떻게 예측하는지 이해하기 위해 연구팀은 AlphaFold 신경망의 각 블록이 구조 예측 과정에서 어떤 역할을 하는지 분석했습니다. AlphaFold의 예측 과정은 매 단계에서 점진적으로 정확한 구조를 형성해 나가며, 특히 어려운 단백질 서열에 대해서도 시간이 지남에 따라 구조가 개선되는 것을 확인할 수 있었습니다.
AlphaFold는 처음 몇 개의 신경망 블록에서 비교적 빠르게 구조를 형성하기 시작하지만, 복잡한 단백질의 경우는 더 많은 신경망 블록을 거쳐야 최종 구조에 도달하게 됩니다. 예를 들어, SARS-CoV-2의 ORF8 단백질의 경우, AlphaFold는 여러 번 구조를 재배열하며, 네트워크가 점진적으로 더 나은 구조를 찾는 과정을 거쳤습니다.
14. 구조 예측 정확도를 높이는 구성 요소
AlphaFold는 여러 기법을 결합해 예측 정확도를 높였습니다. 주요 구성 요소 중 일부를 제거한 실험을 통해, 각 요소가 AlphaFold의 정확성에 얼마나 중요한지 분석했습니다. 결과적으로 Invariant Point Attention (IPA), 재귀적 학습, 그리고 MSA 기반의 보조 목표가 예측 정확도에 중요한 기여를 한다는 것을 확인했습니다.
15. MSA 깊이와 교차 체인 접촉
AlphaFold의 예측 정확도는 MSA의 깊이에 따라 크게 달라집니다. 일반적으로 MSA에 포함된 서열이 많을수록 정확도가 높아지며, 특히 MSA에 포함된 서열이 30개 이하일 때 예측 정확도가 급격히 떨어집니다. 반대로, 서열이 100개 이상일 때는 추가적인 이득이 작아지지만, 서열이 많을수록 여전히 소폭의 개선이 이루어집니다.
또한, AlphaFold는 단일 체인 단백질에 대해 높은 정확도를 보여주지만, 다중 체인 단백질이나 서로 다른 단백질이 복합체를 이루는 경우, 예측 정확도가 떨어지는 경향이 있습니다. 이는 AlphaFold가 단백질의 내부 접촉에 더 의존하기 때문입니다. 향후에는 이러한 다중 체인 단백질의 구조 예측을 개선하기 위한 모델이 필요할 것입니다.
요약
AlphaFold가 단백질 구조를 예측하는 과정은 심층 학습(Deep Learning)을 기반으로, 단백질 서열 정보를 입력받아 3차원 구조를 예측하는 매우 복잡하고 정교한 방식입니다. 이 과정은 크게 다섯 가지 주요 단계로 나눌 수 있습니다:
1. 입력 준비 (MSA 생성 및 템플릿 검색)
AlphaFold는 단백질 서열을 입력으로 받아, 그 서열이 다른 진화적으로 관련된 단백질들과 어떤 상관관계를 가지는지 분석하는 단계부터 시작합니다.
a. MSA (Multiple Sequence Alignment) 생성
- AlphaFold는 주어진 단백질 서열을 다중 서열 정렬(MSA)에 입력하여, 진화적으로 유사한 다른 단백질 서열들을 수집합니다. 이를 위해 jackhmmer 또는 HHblits 같은 도구를 사용하여, 해당 단백질 서열과 유사한 다른 단백질 서열들을 찾습니다.
- MSA는 같은 위치에서 유사한 아미노산들이 어떤 패턴을 보이는지 정렬해 보여줍니다. 이 정보는 단백질의 공진화 패턴(co-evolution)을 파악하는 데 도움이 됩니다. 즉, 진화 과정에서 함께 변한 아미노산들 간의 상호작용 가능성을 예측할 수 있습니다.
b. 템플릿 검색
- AlphaFold는 PDB (Protein Data Bank)에서 실험적으로 밝혀진 단백질 구조를 검색해, 예측하려는 단백질과 유사한 구조를 가진 템플릿을 찾습니다. 이러한 템플릿 정보는 단백질 구조를 더 정확하게 예측하는 데 도움이 됩니다.
2. 입력 데이터 처리 및 특징 추출
AlphaFold는 MSA 및 템플릿 데이터를 기반으로 다양한 특징을 추출합니다. 이때 핵심적인 역할을 하는 것이 Evoformer 블록입니다.
a. Evoformer를 통한 정보 처리
- Evoformer는 MSA와 잔기 간 상호작용(pairwise interactions)을 처리하여, 단백질 구조를 예측하는 데 필요한 중요한 정보를 추출합니다.
- MSA 처리: MSA에서 진화적으로 보존된 위치, 변이가 일어난 위치 등을 분석하여 단백질 서열에서 중요한 특징들을 추출합니다. 진화적으로 보존된 위치는 단백질의 기능적, 구조적으로 중요한 부위일 가능성이 큽니다.
- 잔기 간 상호작용 처리: 단백질 구조 내에서 아미노산 잔기들 간의 상호작용(예: 거리, 상호작용력)을 처리하여, 3차원 구조에서 각 잔기가 어떻게 배치될지를 예측합니다.
Evoformer는 서열 간 상호작용(세로 축)과 잔기 간 상호작용(가로 축)을 Axial Attention을 통해 처리하여, 단백질 서열과 잔기 간의 복잡한 관계를 학습합니다.
3. 구조 모듈(Structure Module)로 3D 구조 생성
Evoformer에서 처리된 데이터를 바탕으로, AlphaFold는 구조 모듈(Structure Module)을 사용해 단백질의 3차원 좌표를 직접 예측합니다.
a. Invariant Point Attention (IPA)
- IPA는 각 잔기의 3D 좌표를 직접 예측하는데 사용되는 핵심 메커니즘입니다. 잔기 간의 상대적인 거리와 방향을 이용해 잔기들이 단백질 내에서 어떻게 배치될지 예측합니다.
- 각 잔기의 위치는 회전 행렬과 변환 벡터로 표현됩니다. 이 정보는 각 잔기의 위치를 3차원 공간에서 정확하게 예측할 수 있도록 도와줍니다.
b. 구조의 반복적 갱신(Iterative Refinement)
- AlphaFold는 한 번에 구조를 완성하는 것이 아니라, 반복적 학습을 통해 점진적으로 구조를 개선해 나갑니다. 예측된 구조를 다시 네트워크에 입력하여, 더욱 정확한 예측을 할 수 있도록 갱신합니다. 이는 예측이 반복될수록 구조의 정확도가 향상된다는 장점이 있습니다.
4. 구조 평가 및 신뢰도 예측 (pLDDT)
AlphaFold는 예측된 구조의 신뢰도를 함께 제공합니다. 이는 pLDDT (Predicted Local Distance Difference Test)라는 지표로 표현됩니다.
a. pLDDT 신뢰도 점수
- pLDDT는 AlphaFold가 예측한 각 잔기의 신뢰도를 나타내는 점수입니다. 0에서 100 사이의 값을 가지며, 값이 높을수록 예측된 구조가 신뢰할 만하다는 의미입니다.
- pLDDT는 예측된 구조의 각 잔기가 실제 구조에서 얼마나 정확한 위치에 있을지를 예측하는 데 사용됩니다. 일반적으로 pLDDT가 90 이상이면 매우 신뢰할 만한 구조로 간주됩니다.
b. TM-score
- AlphaFold는 또한 TM-score를 통해 예측된 전체 구조와 실제 실험적으로 밝혀진 구조 간의 유사성을 평가합니다. TM-score는 0에서 1 사이의 값을 가지며, 값이 1에 가까울수록 두 구조가 유사하다는 것을 의미합니다.
5. 최종 구조 출력 및 후처리
AlphaFold는 예측된 구조를 GPU에서 빠르게 계산해 냅니다. 일반적으로 단백질 서열의 길이에 따라 시간이 달라지며, 수백 개의 잔기를 가진 단백질 구조는 몇 분에서 몇 시간 내에 예측이 완료됩니다.
a. 후처리 및 구조 안정화
- 예측된 구조는 물리적 제약을 고려하여 후처리 과정에서 약간의 수정이 이루어질 수 있습니다. 예를 들어, 일부 결합 각도나 거리 제약을 만족시키기 위해, 구조가 조정될 수 있습니다.
b. 전체 예측 과정의 자동화
- AlphaFold는 처음부터 끝까지 완전히 자동화된 방식으로 동작합니다. 입력 서열을 주기만 하면 MSA 생성부터 최종 구조 예측까지 모든 과정이 자동으로 이루어집니다.
궁금점들
1.단백질 잔기는 정확히 무슨 개념인가
단백질 잔기(Residue)는 단백질을 구성하는 아미노산의 기본 단위입니다. 좀 더 구체적으로 말하면, 단백질은 아미노산들이 연결된 사슬 구조로 이루어져 있는데, 이 아미노산들이 단백질 사슬 내에서 각자 특정 위치를 차지하면서 단백질의 형태와 기능에 기여합니다. 이때 각각의 아미노산을 단백질 사슬에서 '잔기(Residue)'라고 부릅니다.
단백질과 아미노산의 관계
단백질은 많은 아미노산이 펩타이드 결합(peptide bond)으로 연결된 사슬입니다. 아미노산 하나하나가 단백질의 기능을 수행하는 데 중요한 역할을 하며, 단백질 전체의 구조와 기능을 결정합니다.
아미노산은 아미노기(-NH2), 카복실기(-COOH), 그리고 측쇄(R 그룹)로 구성됩니다. 이 측쇄는 아미노산마다 고유한 화학적 성질을 가지며, 단백질의 다양성을 만들어냅니다. 아미노산이 단백질 사슬에 결합되면 물분자가 떨어져 나가고, 그 결과 남은 아미노산을 잔기(Residue)라고 부릅니다.
비유를 들어 설명
단백질을 하나의 긴 기차로 생각해 볼 수 있습니다. 기차는 각 칸이 연결되어 있는 열차로 이루어져 있으며, 각 칸이 기차의 기능을 담당합니다. 이때 각 기차의 칸 하나하나가 아미노산에 해당합니다.
- 열차 칸이 기차에 연결되면: 각각의 아미노산이 단백질 사슬에 결합되는 것처럼, 열차 칸이 기차에 연결됩니다.
- 기차 칸을 잔기(Residue)로 부르는 이유는, 이 칸들이 기차 전체의 기능을 수행하는 데 필수적이기 때문입니다. 단백질에서 각각의 잔기는 단백질 구조와 기능을 수행하는 작은 단위입니다.
기차가 여러 칸으로 이루어져 다양한 기능을 수행하는 것처럼, 단백질도 여러 잔기(아미노산)로 이루어져 그 역할을 합니다.
단백질 잔기의 구조적 역할
단백질에서 각 잔기는 다음과 같은 역할을 합니다:
- 구조: 잔기들은 서로 상호작용하며, 단백질의 3차원 구조를 형성하는 데 기여합니다. 각 잔기의 위치와 상호작용은 단백질의 구조적 안정성에 큰 영향을 미칩니다.
- 기능: 특정 잔기들은 효소의 활성 부위, 단백질-단백질 상호작용 부위 등에 위치하여 단백질의 기능을 직접적으로 수행합니다. 예를 들어, 리신(Lysine)이나 시스테인(Cysteine) 같은 아미노산 잔기들은 효소 활성에 중요한 역할을 할 수 있습니다.
요약
- 단백질에서 잔기는 단백질을 구성하는 아미노산의 단위입니다.
- 아미노산이 단백질 사슬에 결합되어 펩타이드 결합을 형성한 후 남은 부분을 잔기라고 부릅니다.
- 비유하자면, 기차의 각 칸이 단백질의 잔기에 해당하며, 이 잔기들이 모여 단백질의 구조와 기능을 결정합니다.
2.IPA 개념이 attention을 이용해서 예측하고 새로운 잔기를 재귀학습으로 더 학습한다고 했는데 강화학습이랑 비슷한 개념인가? 그리고 기존 attention 네트워크는 학습할때 이런식으로 재귀적 학습을 하지 않는것인가? 딥러닝은 기본적으로 iteration을 통해 업데이트 해서 accuracy를 높이는거 아닌가?
AlphaFold에서 사용되는 IPA(Invariant Point Attention)는 전통적인 강화학습(Reinforcement Learning)과는 약간 다른 방식으로 작동합니다. 또한 일반적인 Attention 메커니즘과도 다소 차별화된 재귀적 학습 방식을 채택하고 있습니다. 당신이 언급한 부분을 바탕으로, IPA와 기존 Attention 메커니즘, 그리고 재귀적 학습의 차이점에 대해 설명하겠습니다.
1. IPA(Invariant Point Attention)와 재귀적 학습
IPA는 단백질의 3차원 구조를 예측하기 위해 개발된 Attention 메커니즘입니다. 이 메커니즘은 주어진 잔기들 사이의 위치적 관계를 통해 단백질의 구조적 제약을 학습합니다.
AlphaFold에서 IPA는 단백질의 잔기 간 3D 공간에서의 상대적 위치(회전과 번역)를 학습하는 데 사용됩니다. 잔기들의 기하학적 관계를 기하학적 불변성(geometric invariance)을 유지하면서 Attention 메커니즘을 통해 예측하고, 그 결과를 다시 모델로 피드백하여 점진적으로 구조를 개선하는 재귀적 학습(iterative refinement) 방식을 사용합니다.
재귀적 학습의 핵심:
- 예측된 구조를 다시 입력으로 사용: AlphaFold는 한 번 예측된 구조를 네트워크로 다시 입력해, 해당 구조를 바탕으로 더 나은 예측을 할 수 있도록 반복합니다. 이 과정을 통해 점점 더 정확한 단백질 구조를 예측할 수 있습니다.
- 이 재귀적 학습은 연속적인 업데이트 과정을 통해 구조를 점점 개선한다는 점에서 일반적인 딥러닝의 학습 방식과 유사하지만, 구체적으로는 출력 결과(구조)를 반복적으로 피드백하여 더 나은 구조를 예측한다는 점에서 차이가 있습니다.
IPA와 강화학습의 차이:
- 강화학습(Reinforcement Learning)에서는 에이전트(agent)가 환경(environment)에서 행동(action)을 수행하고, 그 행동에 대한 보상(reward)을 바탕으로 학습합니다. 이 보상을 극대화하는 방향으로 정책을 최적화하는 것이 목표입니다.
- IPA는 강화학습과 다르게 명시적인 보상(reward)이 없으며, 대신 단백질의 잔기 간 기하학적 관계를 학습하면서 점진적으로 더 정확한 3D 구조를 예측합니다. 따라서 강화학습처럼 정책을 최적화하는 방식이 아니라, 예측된 구조를 반복적으로 개선하는 재귀적 최적화 과정을 통해 구조를 정확하게 맞추는 것이 목표입니다.
2. 기존 Attention 메커니즘과 재귀적 학습의 차이점
Attention 메커니즘은 주로 딥러닝의 자연어 처리(NLP)에서 사용되며, 다양한 입력 요소들 간의 관계를 모델링하는 데 탁월한 성능을 보입니다. 일반적인 Self-Attention 메커니즘은 다음과 같은 과정을 거칩니다:
- 입력 데이터에 대해 Query, Key, Value를 생성합니다.
- Query와 Key의 내적(dot product)을 구해 두 요소 간의 연관성을 계산하고, 이를 기반으로 Value를 가중합(weighted sum)하여 최종 출력을 만듭니다.
이 과정은 한 번에 이루어지며, 일반적으로 재귀적이지 않습니다. 즉, 기존의 Attention 메커니즘은 한 번의 계산 과정에서 모든 입력 간의 상호작용을 모델링하고, 이를 기반으로 출력을 내보냅니다.
재귀적 학습과 기존 Attention의 차이점:
- 기존 Attention 메커니즘: 단일 패스(single pass)로 입력 간 상호작용을 계산하며, 학습은 미리 정의된 에폭(epoch) 내에서 각 입력이 반복적으로 학습됩니다. 하지만, 예측된 출력값을 다시 입력으로 사용해 점진적으로 성능을 개선하는 방식은 사용되지 않습니다.
- AlphaFold의 재귀적 학습: 예측된 구조를 다시 입력으로 사용하여, 그 구조를 더 개선하기 위해 다시 학습하는 과정을 거칩니다. 즉, 예측된 결과물이 다시 네트워크에 피드백되며, 이를 통해 점진적 구조 개선이 이루어집니다. 이는 기존의 Attention 네트워크에서 볼 수 없는 재귀적 학습 방식입니다.
3. 딥러닝의 기본 학습 방식과 재귀적 학습의 차이
딥러닝 모델은 기본적으로 에폭(epoch) 단위로 데이터셋을 여러 번 반복 학습하여 성능을 점진적으로 향상시킵니다. 이 과정은 백프로파게이션(backpropagation)을 통해 가중치를 업데이트하고, 미니배치(minibatch)나 스텝(step) 단위로 손실(loss)을 줄여 나가는 방식입니다.
그러나 AlphaFold의 재귀적 학습 방식은 단순히 여러 에폭 동안 학습하는 것과는 다릅니다. 재귀적 학습은 모델의 예측값(단백질 구조)이 모델의 다음 입력으로 다시 사용된다는 점에서 다릅니다. 이렇게 하면 초기 구조가 잘못되었을 경우, 이를 수정하면서 점점 더 정확한 예측으로 수렴할 수 있습니다.
4. 재귀적 학습의 장점
- 구조 개선: AlphaFold에서 재귀적 학습은 초기 예측된 구조가 부정확하더라도, 이를 반복적으로 수정해 점진적으로 정확한 구조에 가까워지게 만듭니다.
- 고차원 정보 통합: 여러 번의 재귀를 거치면서 단백질의 잔기 간 기하학적 제약과 공진화 패턴이 더 잘 반영된 최종 구조를 예측할 수 있습니다.
요약:
- IPA는 기하학적 불변성을 가진 Attention 메커니즘으로, 잔기 간 3차원 공간에서의 관계를 학습합니다. 이는 강화학습과 달리, 보상 기반이 아닌 구조적 관계를 점진적으로 개선하는 방식입니다.
- 재귀적 학습은 기존의 Attention 메커니즘과 달리, 예측된 결과를 다시 입력으로 사용하여 구조를 반복적으로 개선하는 방식입니다.
- 딥러닝의 기본 학습 방식은 데이터를 여러 번 반복 학습하면서 가중치를 점진적으로 업데이트하는 방식인 반면, AlphaFold는 구조 자체를 반복적으로 개선하는 재귀적 학습 방식을 채택하여 더욱 정교한 예측을 수행합니다.
