0. Introduction
생명정보학(Bioinformatics)이 생물학 연구에 가장 크게 공헌한 점은 DNA 혹은 protein sequence의 기능을 밝힌 것이다. 많은 생명정보학 알고리즘 중 Sequence Alignment는 기존에 기능이 밝혀진 DNA 또는 protein sequence와 기능이 밝혀지지 않은 sequence의 유사도를 계산하여 DNA, protein의 기능 또는 공통조상을 찾는 것을 목적으로 한다. 본 포스트에서는 Sequence Alignment의 정의와 관련된 알고리즘에 대해 작성하고자 한다.
1. Background
Sequence Alignment 설명에 앞서 알아두면 좋을 개념들에 대해 소개한다.
- Homologs
- 공통된 조상을 가지면서 비슷한 DNA sequence를 가지는 종들- Orthlogs: 다른 종임에도 불구하고 공통된 조상으로부터 종 분화(species divergence)로 인해 유사한 DNA sequence를 가지는 경우
- Paralogs: 같은 종에서 gene duplication으로 인해 유사한 DNA sequence를 가짐에도 서로 기능이 다른 경우
- Analogs
- 공통된 조상이 없는데도 불구하고 비슷한 DNA sequence를 가지는 종들
2. Sequence Alignment
Sequence Alignment는 두개 또는 그 이상의 protein sequence가 주어졌을 때, 서로간의 유사도를 계산하고 이를 정렬하는 방법이다. 위에서 언급했듯이 Sequence Alignment는 기능이 알려진 DNA, protein의 sequence와 기능이 알려지지 않은 sequence의 유사도를 계산하여 기능을 유추하거나 공통의 조상을 찾는데 활용된다. 물론 두 sequence가 유사도가 높다고 하더라도 Analogs의 경우처럼 전혀 상관없을 수도 있다. 따라서 Sequence Alignment의 결과는 '두 sequeunce 간 유사도가 높으니 공통된 조상 또는 기능이 비슷하지 않을까?'라는 가설을 세우는데 사용될 수 있을 것이다.
그렇다면 어떻게 두 sequence의 유사도를 측정할 수 있을까? 첫번째로 가장 간단하게 생각할 수 있는 유사도 측정 방법은 두 sequence에서 동일한 위치, 동일한 문자를 가지는 경우의 갯수를 세는 것이다. 예를 들어 ACGTTAT, TCGTACT라는 두 sequence가 있다고 가정하자. 첫번째 유사도 측정 방법을 활용하면 다음과 같다.
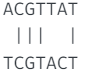
두 sequence는 2번째 C, 3번째 G, 4번째 T, 7번째 T가 동일한 위치, 동일한 문자를 가지는 것을 확인할 수 있다. 하지만 이 방식은 적절하지 않다고 볼 수 있다. 생물은 진화과정을 거치면서 돌연변이(mutation), 삽입(Insertion), 결실(Deletion) 등 DNA, protein의 변화가 일어나게 되는데 첫번째 방식은 이러한 현상들을 반영하지 못하는 것으로 볼 수 있다.
이러한 문제를 해결하기 위해 Sequence Alignment의 유사도 측정 방식은 sequence들의 순서는 유지하며 각 sequence가 공유하는 문자를 찾아 정렬하는 방식을 사용한다. 두번째 방식으로 ACGTTAT, TCGTACT의 유사도를 측정하면 다음과 같다.

두 sequence는 2번째 C, 3번째 G, 4번째 T, 6번째 A, 8번째 T가 동일한 위치, 동일한 문자를 가지는 것을 확인할 수 있다. sequence에 추가된 '-'는 공백문자(gap)이며 Sequence Alignment의 유사도 측정 방법을 활용하기 위한 문자이며 삽입 또는 결실을 의미한다.
2-1.Global Sequence Alignment
Global Sequence Alignment는 주어진 두 개의 sequence의 전체 sequence에 대해 가장 최적의 정렬을 수행한다. 일반적으로 비교하고자 하는 서열의 길이가 비슷하고 유사성이 높은 경우에 사용된다고 한다.
특정 scoring scheme이 주어졌을 때 score는 다음과 같다.
위의 Score를 활용하여 유사도를 계산한다고 하였을 때 x=ATGTTAT, y=ATCGTAC 두 sequence에 대해 Global Sequence Alignment 수행한다고 가정하자.
Exhaustive Search 알고리즘으로 해당 문제를 해결하면 정확한 답을 얻을 수 있지만 계산복잡도가 크다는 단점이 있다. 예시의 sequence는 매우 짧지만 실제 DNA, protein sequence는 길이가 길기때문에 계산복잡도가 높은 알고리즘은 적절하지 못하다. Greedy Algorithm의 경우 계산복잡도가 작지만 결과가 최적의 답이 아닐 가능성이 있다.
따라서 적당한 계산복잡도를 가지며 정확한 답을 얻을 수 있는 Dynamic Programing을 활용하여 Global Sequence를 해결하고자 한다. Dynamic Programming을 위한 Recursive Formula는 다음과 같다.
위의 Recursive Formula는 각 상황(gap, mismatch, match)에 따른 값중 가장 큰 값을 쓰겠다는 의미이다. Global Sequence Alignment의 Pseudo code는 다음과 같다.
GlobalAlignment(x, y)
S[0,0] <- 0
for i <- 1 to m
S[i,0] <- S[i-1,0]-σ
for j <- 1 to n
S[0,j] <- S[0,j-1]-σ
for i <- 1 to m
for j <- 1 to n
if x_i = y_i
S[i,j] = max(S[i-1,j]-σ, S[i,j-1]-σ, S[i-1,j-1] + α)
else
S[i,j] = max(S[i-1,j]-σ, S[i,j-1]-σ, S[i-1,j-1] - μ)
return S[m,n]x="ATGTTAT", y="ATCGTAC", 라고 했을 때 위 Recursive Formula와 Pseudo code를 그림으로 표현하면 다음과 같다.
 (차후 변경 예정)
(차후 변경 예정)
Global Sequence Alignment 결과 두 sequence의 유사도는 11인것을 확인할 수 있다. 최종적인 정렬결과는 Backtracking을 통해 확인할 수 있으며 Backtracking은 Global Sequence Alignment 알고리즘이 완료되면 가장 마지막 값부터 중 가장 큰 점수를 찾아 문자 또는 gap을 출력하는 알고리즘이다.
위의 예제에서는 계산을 간단히 수행하기 위해 penalty, premium값을 임의로 정하였지만 실제 Sequence Alignment를 수행할 때는 PAM(Point Accepted Mutations), BLOSUM(Block Substitution Matrix)같은 Scoring Matrix를 활용한다.
2-2.Local Sequence Alignment
3. Gap penalty
4. Multiple Sequnce Alignment
참조
Introduction to Bioinformatics(2020-2, Graduate course), Young-Rae Cho, Yonsei Univ.
Bioinformatics Algorithms Design and Implementation in Python, Miguel Rocha · Pedro G. Ferreira
Alignment, http://www.incodom.kr/Alignment
