
Rosalind (revp) 문제
A DNA string is a reverse palindrome if it is equal to its reverse complement. For instance, GCATGC is a reverse palindrome because its reverse complement is GCATGC. See Figure 2.
Given: A DNA string of length at most 1 kbp in FASTA format.
Return: The position and length of every reverse palindrome in the string having length between 4 and 12. You may return these pairs in any order.
문제 해설
DNA seq 로부터 palindrom을 찾는 문제입니다. 단순한 Palindrom을 생각한다면 이런 걸 생각했을 지도 모르겠습니다. 예를 들어 "토마토". 앞으로 읽어도 토마토이고 거꾸로 읽어도 토마토이니 palindrom이라고 할 수 있죠. 하지만 DNA에서의 팰린드롬은 조금 다릅니다. 위에서 예시를 들어줬듯이 GCATGC의 경우 오른쪽에서 왼쪽으로 읽는 것과 왼쪽에서 오른쪽으로 읽는 것과는 차이가 있습니다.
- GCATGC -> CGTACG
하지만 중요한 점은 DNA helix에서 3`과 5` 은 서로 반대로 읽어야 함으로
- CGTACG -> GCATGC
위와 같이 변하여 원래 예제인 GCATGC 와 동일하게 변하게 됩니다.
이를 DNA 펠린드롬이라고 합니다.
주어진 변수
- Given : DNA seq가 주어집니다.
>Rosalind_24
TCAATGCATGCGGGTCTATATGCAT - Result : 아래와 같이 DNA 펠린드롬이 시작하는 위치와(1부터 시작) 길이를 표시하면 됩니다.
4 6
5 4
6 6
7 4
17 4
18 4
20 6
21 4
예를 들자면 4번째 시퀀스는 총 6개의 ATGCAT nucleotide로 이루어져 있습니다. 이걸 반대로 TACGTA 로 바꾸고 거꾸로 뒤집으면
ATGCAT == ATGCAT로 같아지게 되고, 이는 곧 펠린드롬이라는 의미입니다.
Solution(2021-08-11 Solved)
- input_processor
들어온 데이터를 처리합니다. fasta file은 시퀀스가 한줄로 되어 있지 않고 여러줄로 되어 있어\n로 잘라주어야 합니다.input_processor = lambda i: i.strip().split("\n") - get_dna_seq
들여쓰기로 쪼개진 리스트에서 DNA 시퀀스만 골라서 한줄로 붙입니다. 시퀀스의 첫번째 줄인 시퀀스 아이디는 제거해 줍니다.
get_dna_seq = lambda lines: ''.join([line for line in lines if not line.startswith(">")])- get_all_cases
DNA 시퀀스를 쪼개서 가장 긴 줄부터 가장 작은 단위까지 모든 가능한 경우의 수를 구합니다. 약간 무식한 방법이지만 데이터를 쪼개는 방식을 잘 하면 1kbp 이상인 시퀀스도 순식간에 풀어낼 수 있습니다.
쪼개는 방식은 재귀를 이용하고 아래와 같은 방식으로 경우의 수들을 찾아냅니다.
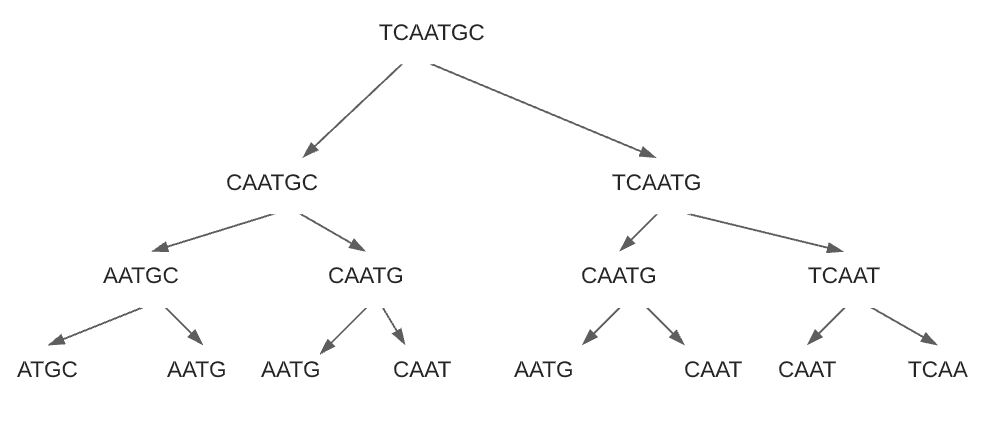
왼쪽 인덱스와 오른쪽 인덱스를 주고 왼쪽으로 갈때는 왼쪽 인덱스 +1, 오른쪽으로 갈때는 오른쪽 인덱스 -1 을 해주면 모든 경우의 수를 구할 수 있습니다.
실제 코드는 아래와 같습니다.(python 허접이라 좀 깁니다.)
def get_all_cases(dna_seq):
print(f"{get_all_cases.__name__} is running")
global cases, idxes, length, memory # 글로벌 변수를 선언해야 재귀 함수에서 리스트나 셋이 리셋되지 않습니다.
memory = set() # 다이나믹 프로그래밍
cases = list() # 경우의 수들
idxes = list() # DNA 시퀀스에서 시작 인덱스
length = list() # 경우의 수의 DNA 서열 길이
# 위에서 말했던 재귀함수가 여기서 돌아갑니다.
def seq_tree(s, left_idx, right_idx):
# 오른쪽과 왼쪽 인덱스가 만나면 종료
if right_idx - left_idx == 0:
return
# 다이나믹 프로그래밍. memory에 있으면 종료
if s[left_idx:right_idx] in memory:
return
else:
memory.add(s[left_idx:right_idx]) # 메모리에 저장
cases.append(s[left_idx:right_idx]) # 경우의 수 저장
idxes.append(left_idx) # 시작 인덱스 저장
length.append(len(s[left_idx:right_idx])) # 경우의 수 길이 저장
seq_tree(s, left_idx + 1, right_idx) # 왼쪽으로
seq_tree(s, left_idx, right_idx - 1) # 오른쪽으로
# 실행하면 글로벌 변수로 선언했던 배열들이 모든 경우의 수와 관련 메타데이터를 기억하고 있습니다.
seq_tree(dna_seq, 0, len(dna_seq))
# 원본 DNA 시퀀스 데이터와 경우의수 및 메타데이터를 반환
return dna_seq, zip(idxes, cases, length)- filter_seq
들어온 데이터를 필터링 합니다. 문제에서 길이가 4 이상 12 이하여야 한다고 했고, DNA 팰린드롬은 중간에 홀수면 불가능함으로(ATC -> TAG) 반드시 길이가 2의 배수여야 합니다.
def filter_seq(data_pacakge):
dna_seq, cases_zip = data_pacakge
cases_zip = map(lambda x:(x[0], x[1]), filter(lambda x: 4<=x[2]<=12 and x[2] % 2 == 0, cases_zip))
return dna_seq, cases_zip- get_pelindrom
이제 팰린드롬을 구할 준비가 되었습니다. 팰린드롬을 구하는 방법은 간단합니다. 필터링된 경우의 수들을 상보적 DNA로 바꾼 뒤에 거꾸로 뒤집습니다. 그런다음 원본 경우의 수와 변환한 DNA 서열을 비교해서 같으면 팰린드롬이라 판단하고 시작 인덱스와 길이를 튜플로 배열에 담는 것입니다.
def get_pelindrom(data_package):
print(f"{get_pelindrom.__name__} is running")
dna_seq, cases_zip = data_package # 필터링 된 데이터
palindroms = []
for idx, dna_frag in cases_zip:
matches = re.finditer(get_reverse_dna(dna_frag), dna_seq) # 실제 데이터에서는 같은 경우의 수가 원본 시퀀스에서 여러군데 발견될 수 있습니다.
match_idxes = [match.start() for match in matches] # 시작인덱스를 구합니다.
for match_idx in match_idxes:
if is_palindrom(dna_frag): # 펠린드롬인지 확인합니다.
palindroms.append((match_idx + 1, len(dna_frag))) # 펠린드롬이 맞다면 배열에 추가합니다.
return palindroms- result_print
튜플로된 시작과 길이 데이터 리스트를 출력합니다.
def result_print(palindroms):
for start_idx, length in palindroms:
print(start_idx, length)- 파이프라인에 담고 돌립니다!
# compose는 toolz의 compose 함수를 사용하였습니다.
run = tz.compose(
result_print,
tz.curry(sorted, key=lambda x: x[0]),
get_pelindrom,
filter_seq,
get_all_cases,
get_dna_seq,
input_processor
)
run(test_dna_seq)전체 코드
import regex as re
import toolz as tz
test_dna_seq = """
>Rosalind_24
TCAATGCATGCGGGTCTATATGCAT
"""
compl_dna = {
"A": "T",
"G": "C",
"C": "G",
"T": "A"
}
input_processor = lambda i: i.strip().split("\n")
get_dna_seq = lambda lines: ''.join([line for line in lines if not line.startswith(">")])
get_reverse_dna = lambda dna_seq: "".join(reversed([compl_dna[n] for n in dna_seq]))
def filter_seq(data_pacakge):
dna_seq, cases_zip = data_pacakge
cases_zip = map(lambda x:(x[0], x[1]), filter(lambda x: 4<=x[2]<=12 and x[2] % 2 == 0, cases_zip))
return dna_seq, cases_zip
def get_all_cases(dna_seq):
global cases, idxes, length, memory
memory = set()
cases = list()
idxes = list()
length = list()
def seq_tree(s, left_idx, right_idx):
if right_idx - left_idx == 0:
return
if s[left_idx:right_idx] in memory:
return
else:
memory.add(s[left_idx:right_idx])
cases.append(s[left_idx:right_idx])
idxes.append(left_idx)
length.append(len(s[left_idx:right_idx]))
seq_tree(s, left_idx + 1, right_idx)
seq_tree(s, left_idx, right_idx - 1)
seq_tree(dna_seq, 0, len(dna_seq))
return dna_seq, zip(idxes, list(cases), length)
def is_palindrom(dna_seq):
rev_seq = get_reverse_dna(dna_seq)
if rev_seq == dna_seq:
return True
return False
def get_pelindrom(data_package):
dna_seq, cases_zip = data_package
palindroms = []
for idx, dna_frag in cases_zip:
matches = re.finditer(get_reverse_dna(dna_frag), dna_seq, overlapped=False)
match_idxes = [match.start() for match in matches]
for match_idx in match_idxes:
if is_palindrom(dna_frag):
palindroms.append((match_idx + 1, len(dna_frag)))
return palindroms
def result_print(palindroms):
for start_idx, length in palindroms:
print(start_idx, length)
run = tz.compose(result_print, tz.curry(sorted, key=lambda x: x[0]), get_pelindrom, filter_seq, get_all_cases, get_dna_seq, input_processor)
run(test_dna_seq)
Bioinformatician