Bioinformatics_Pairwise-Alignments_Dynamic-Programming(Alignments_Algorithm)
Bioinformtics 분야에서 Pairwise Alignment를 위해 여러 알고리즘이 사용되는데 그 중 가장 효율적인 알고리즘 중 하나가 Dynamic Programming 이다.
Pairwise Alignment는 두 개의 SequencePairwise alignment가 있을 때 두 Sequence간의 similar한 region을 찾거나 어떠한 한 Sequence가 어디로 부터 온 것인지, 가장 유사한 Sequence가 무엇인지 찾기 위한 과정이다. (전혀 다른 말도 안되는 걸 하지는 않고 상대적으로 유사한 sequence를 갖고 한다고 한다.)
DB에서 BLAST라는 툴을 이용해서 simiral한 Sequence를 찾는 것이고 multiple sequence alignment의 기본이다.
Alignment는 두 가지 factor에 의해 결정 된다.
-
Alignment Algorithm
-
Alignment Scoring (parameter)
최장부수열만을 구하기 위한 일반적인 알고리즘에서 다루는 LCS 문제와 달 best alignment를 찾기 위해 Sequence 내의 gap도 고려하게 된다.
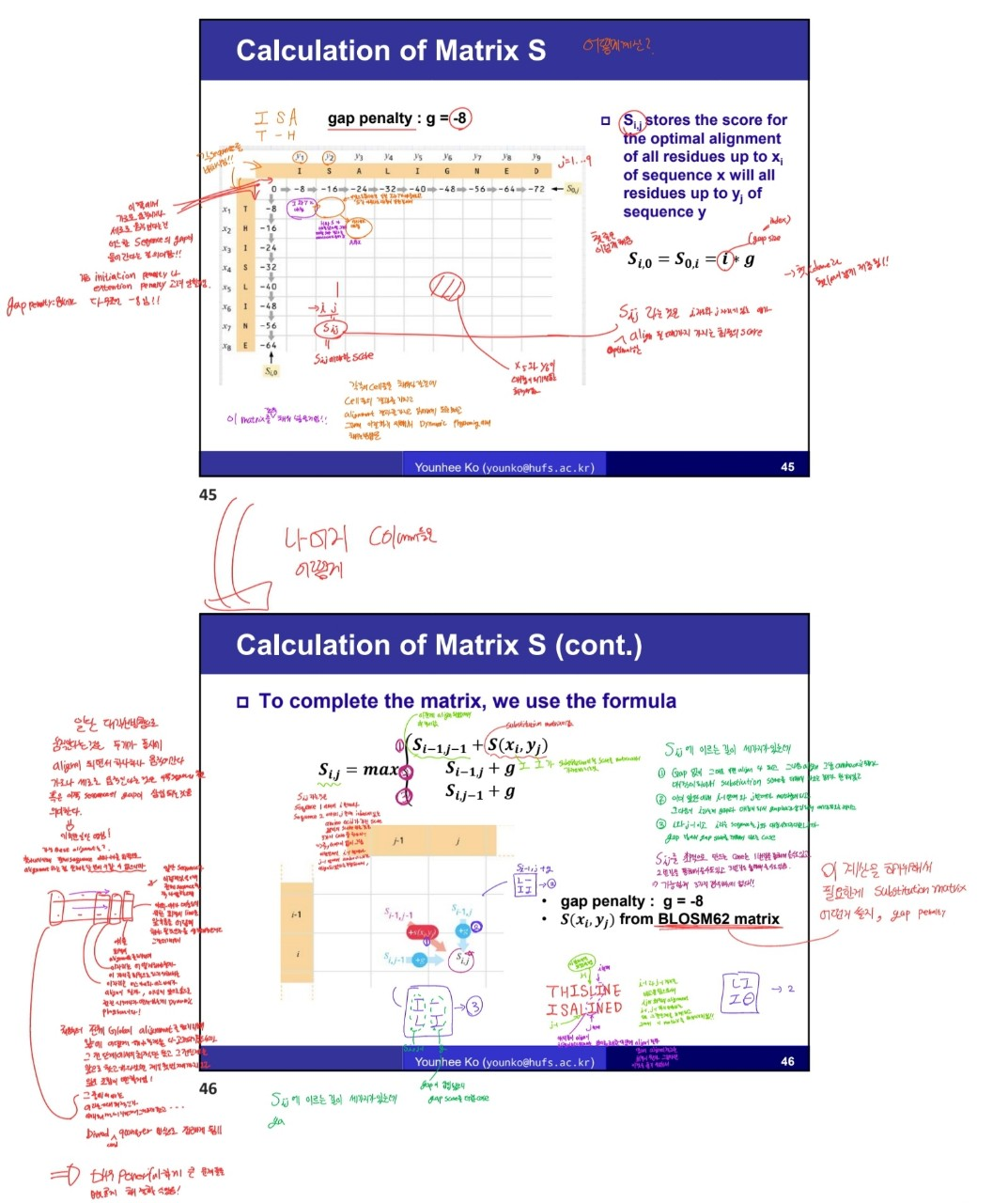
Sequence 전체에 대해서 alignment하고자하는 Global Aligment와 가장 높은 level의 유사도를 가지는 local reigion을 찾기 위한 Local Alignment가 있다. 이들 중 Global Pairwise Alignment 알고리즘 들 중에 가장 대표적인 Needleman-Wunsch 알고리즘을 통해 Alignment하는 방법은 다음과 같다.

1. Matrix S를 정의하고 원소 값들을 계산하여 전체 Matrix의 원소를 모두 채워 넣는다.
2. Global Alignment를 오른쪽 corner 아래로 부터 왼쪽 상부로 올라가면서 읽어가며 찾는다.
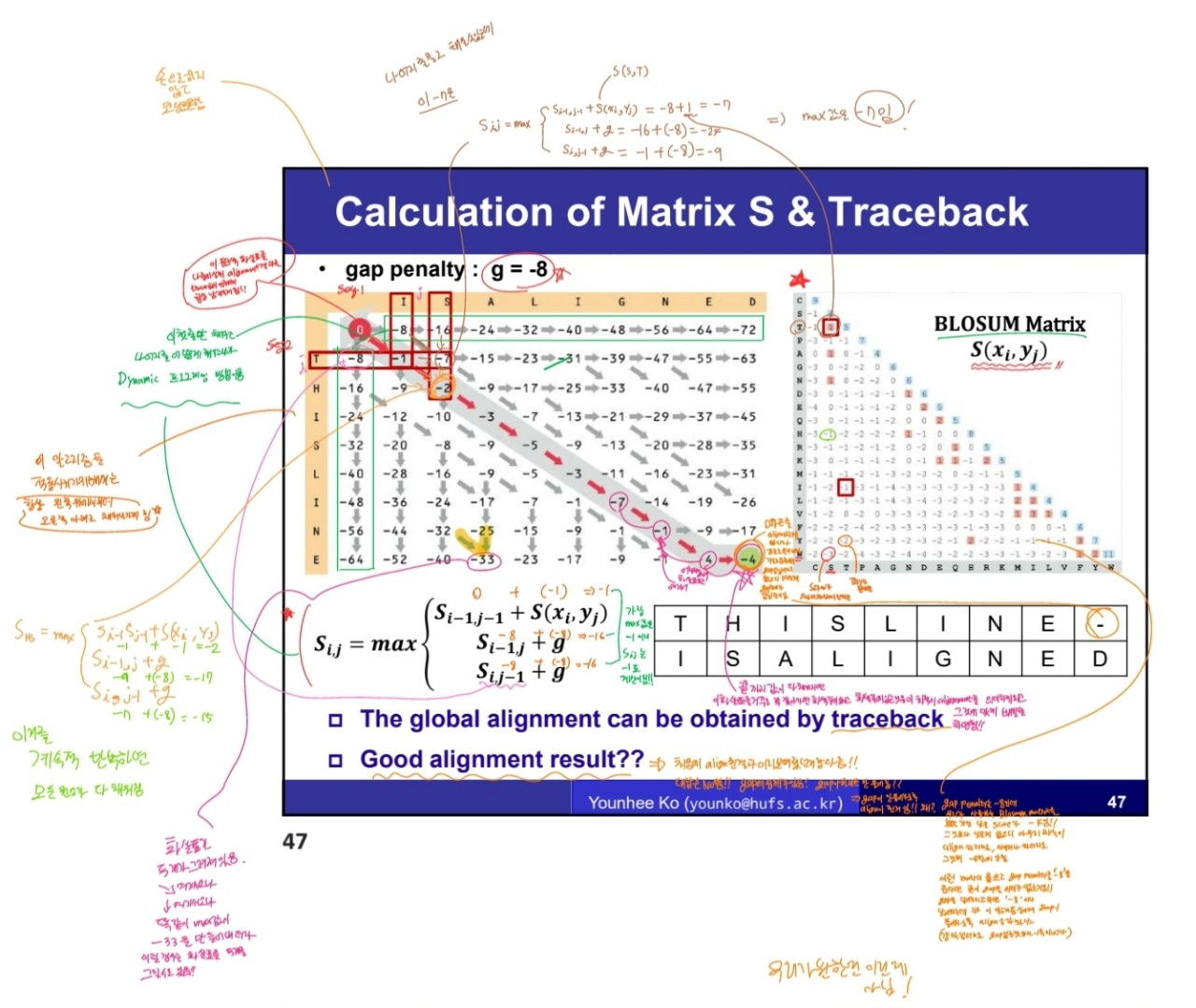
But, 이것은 Good alignment 결과는 아니다.
Needleman-Wunsch 알고리즘은 두 Sequence에 대한 Alignment를 위해 Substitution Matrix와 gap penalty를 이용한다.
여기서 gap에 문제가 있다. 위 그림에 있는 결과는 gap이 안 들어가도록 align 된 것이다. 왜냐하면 gap penalty가 -8로 설정되어 있는데 Substitution Matrix로 사용하는 Blosum Matrix를 보면 가장 낮은 score가 -4다. -4보다 낮은 원소 값이 없으니 아무리 최악의 align이라 하더라도 -4밖에 안 되는 것이다. 즉, 이러한 Matrix를 사용면 막 넣어도 gap을 넣는 것보단 나은 결과가 되니 굳이 gap의 의미가 없어지게 되는 것이다.
중요한 점은 Sequence가 매우 close하거나 divergent하다고 할 때 그것에 따라 적당한 Blosum matrix를 결정하게 되고 그 Blosum Matrix에 있는 score들의 분포를 보고 그것에 맞게 gap penalty를 조정해야 한다는 것이다.
정답은 없고 상황에 맞게 gap penalty를 조정해야된다. 어떻게 하느냐에 따라 결과가 달라질 수 있다!
그럼 위 문제점을 개선하기 위해 gap penalty를 -4로 변경해보면 다음과 같은 결과가 나온다.

지금까지는 Protein Sequence 를 가지고 Alginment 한 것이고 DNA Sequence에 적용해보면 다음과 같다.
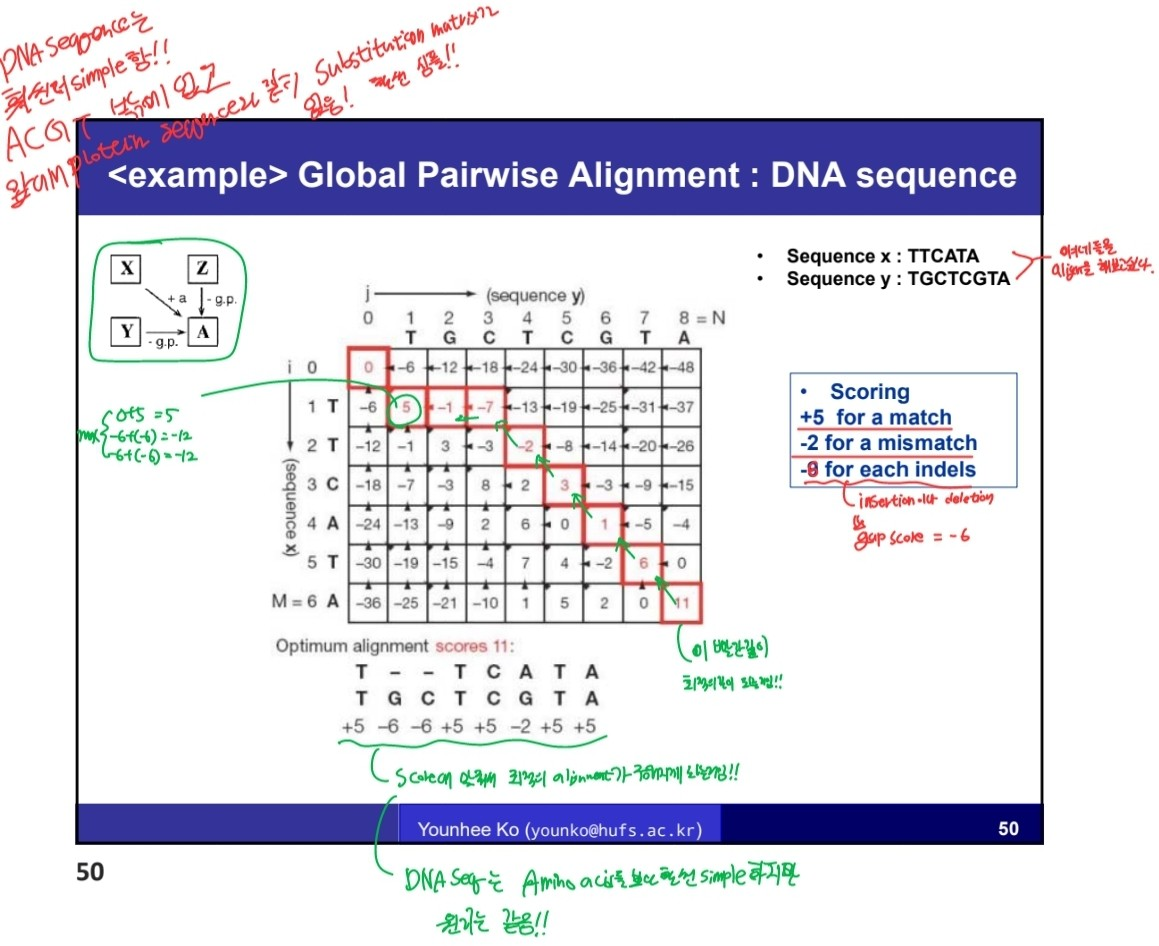
.
