
DeepDTA 는 Drug Target Interaction(DTI) 분야에서 가장 많이 인용되고, 새로운 모델을 만든 뒤, 비교하는 Baseline model 로 항상 등장하는 모델이다.
1. Summary
목표 :
딥러닝 기반 모델을 사용하여 오직 Protein 시퀀스 와 Drug Smiles 정보만을 사용하여 'Binding Affinity'를 예측해보자!! Regression Problem
기존 2D, 3D 기반 연구가 진행되었지만 위 논문에서는 1D CNN을 활용
Dataset :
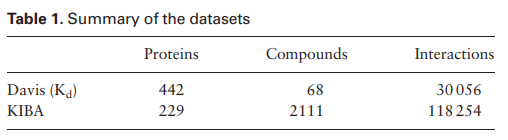
Davis & KIBA
Input :
[Drug] SMILES --> int ex. [C N = C = O]=>[1 3 63 1 63 5]
[Protein] Seq.
둘다 너무 길어서 최대 길이를 SMILES - 85 / Protein - 1200 로 고정(Davis기준)
SMILES - 100 / Protein - 1000
남는 부분은 '0'으로 padding
아무리 단백질의 80% 커버하고 / compound의 90% 커버한다고 해도
※이게 문제가 되지는 않을까??
Model :
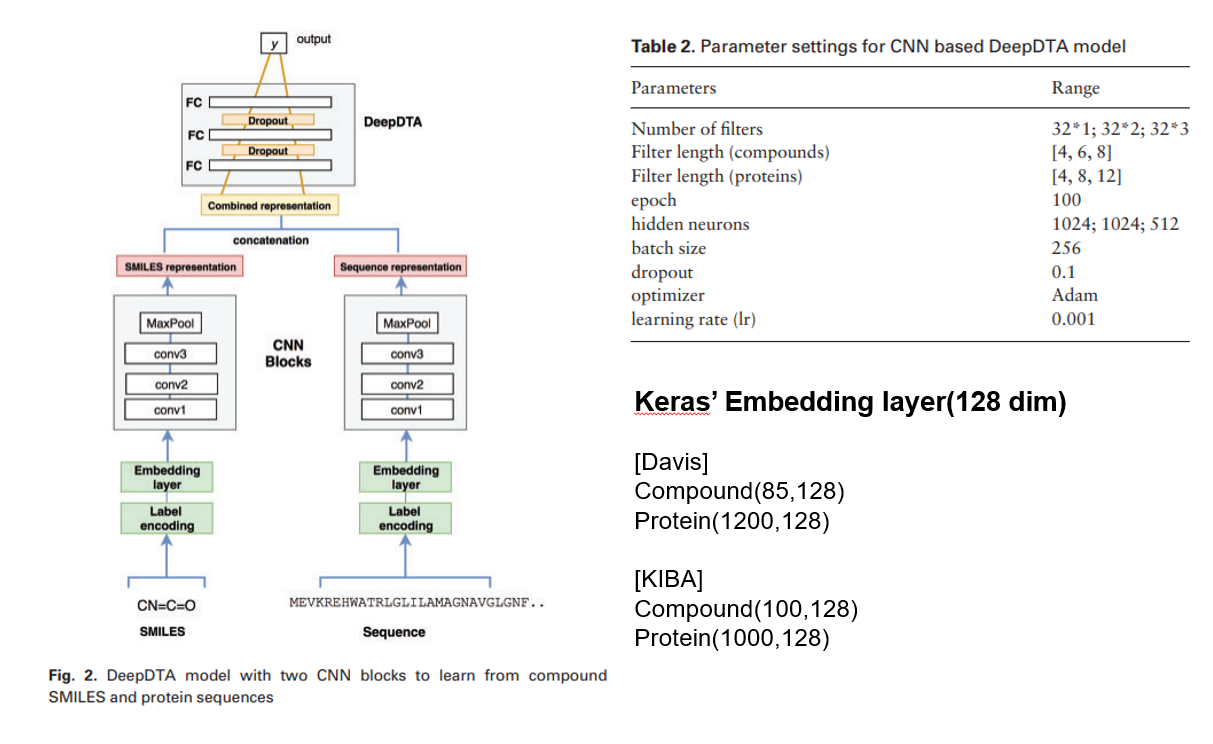
Baseline :
Compound - Pubchem Sim --> compound similarity
Protein - Smith-Waterman algorithm --> protein similarity
1. Kron-RLS Pahikkala et al., 2014
2. Simboost He et al., 2017
- To predict BA scores with a gradient boosting machine
둘다 Traditional machine learning algorithms 이용하고 2D-Representation
Result :
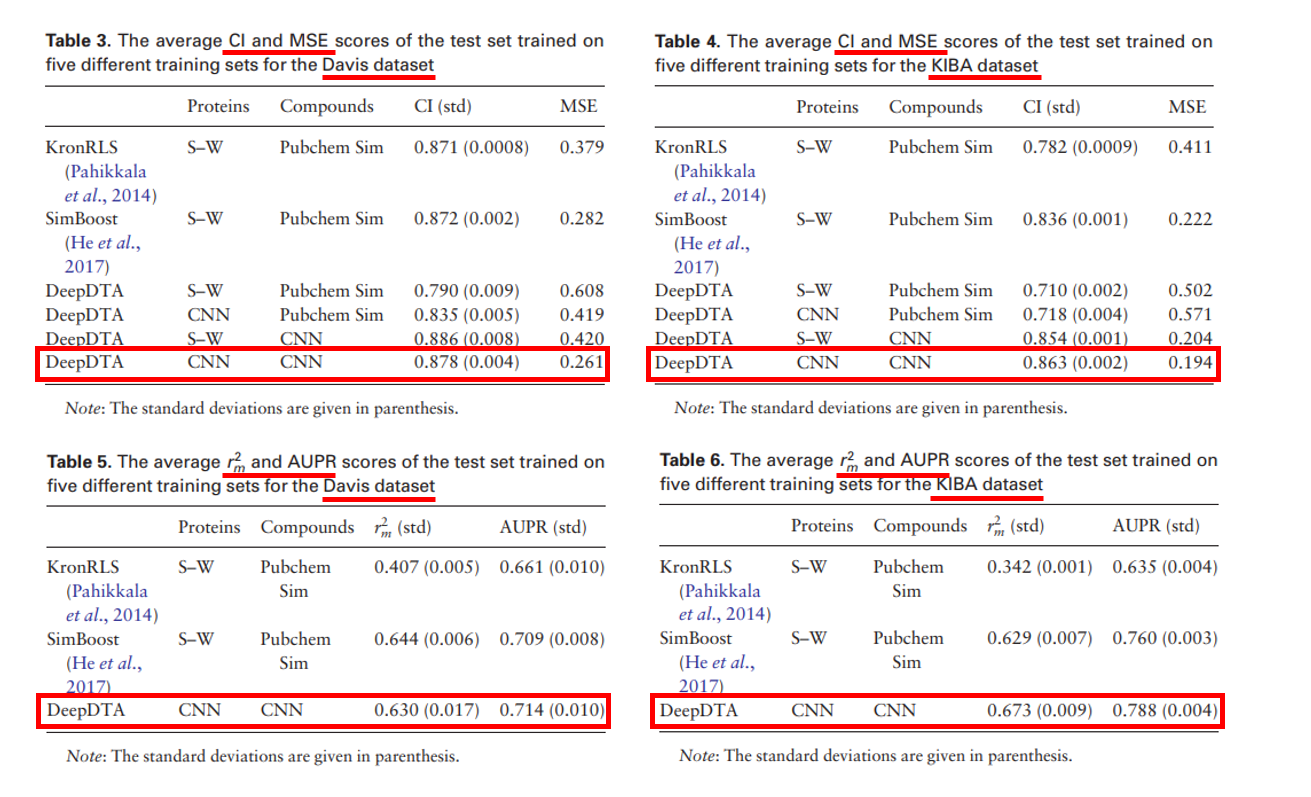 1) CI : Concordance index 관련논문
1) CI : Concordance index 관련논문
예측모델이 얼마나 좋은지 평가하는 지표
2) MSE : Mean Sqaured Error
Regression 모델에서 가장 흔히 쓰는 값
예측값과 실제값 간의 차를 제곱한 값
당연히 MSE 값이 낮아야 성능이 좋다!
3) rm^2 관련논문
QSAR 모델 성능 평가 지표
rm^2 값이 0.5 보다 높으면 "ACCEPTABLE"
4) AUPR : (Area Under Precision Recall)
Binary prediction 연구에 주로 사용
위 논문에서는 AUPR을 측정하기 위해 Binding Affinity를 기준으로 Threshold를 설정하여 Binary 형태로 변환시켰다.
Davis에서는 pKd 7
KIBA에서는 KIBA 12.1
2. 궁금한 점
2-1.
위에서 말한대로 오직 시퀀스 데이터만 이용해서 딥러닝을 돌리는데 가장 중요한 smiles 와 protein seq를 다 사용하지 않고 input으로 사용해도 문제가 없는가? (너무 결과만 나오는것에 급급한 것은 아닌가?)
2-2.
개인적으로 DTI에서는 단백질 구조와 chemical 구조의 상호작용이 큰 역할을 한다고 생각한다. 오직 시퀀스로만 가지고 예측을 한다고하면 현실성이 너무 없지 않은가?
-> Introduction을 보면 기존의 CNN을 이용하여 protein-ligand complex(3D)로 접근해보았지만, 알려진 결합이 25,000개 밖에 되지 않아 한계가 있다. 그래서 시퀀스로 일단 접근해본것 같다.
