tag: 2-4 길이의 amino acid sequence. PTM identification을 할 때 유용하게 쓰임
dp algorithm을 통해서 database에 있는 펩타이드와 일치하는 sequence tag의 modification을 identify
MODa scoring algorithm
multiple tags를 기반으로 한 dynamic programming을 쓴다.
- S: MS/MS Spectrum
- P: a1...an -> DB에서 선택된 펩타이드
- T: set of tags matched to P
길이가 n인 tag는 n+1의 질량들로 define된다. -> 무슨 의미이지
start(t): tag t의 시작 위치(시작하기 직전 위치가 더 맞는듯..?)
end(t): tag t의 종료 위치
(start(t) = 0, end(t) = 1이라면 a1만 tag임을 의미한다.)
M[p][t][s]: maximum score path at position p.
여기서 s는 대각선 위치가 tag의 before인지, after인지를 나타낸다. (0: before, 1: after)
이 third dimension s는 각각의 tag를 여러 개의 subtags로 나누지 않게 하기 위함이다.
As defined, one can jump inside a tag at any point and jump out again as soon as at least one amino acid is used in the tag(branch labeled as 'jumps inside the tag')
-> 어느 지점에서든지 tag안으로 바로 점프 가능. 그리고 tag 안에서 적어도 아미노산 하나가 사용이 되었다면 바로 tag 밖으로 점프 가능
-> 무슨 의미인지 더 생각해봐야겠다.
M[p][t][s]는 0으로 초기화가 된다. score(p,t)가 아래 규칙에 재귀적으로 더해진다.
-
Amino acid jumps
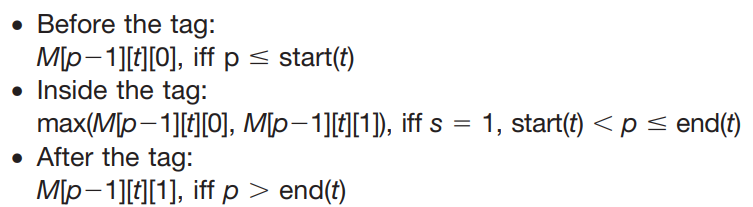
-
Modification jump
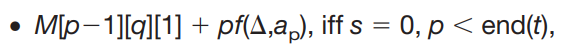
over all tags q such that start(q) < start(t), where pf(d, aa) <= 0 is a penalty function for a modification d on the amino acid(aa).
pf(d, aa) <= 0는 a라는 아미노산에 대한 modification에 대한 패널티이다.
여기서 d는 mass의 displacement를 의미하며, modification에 의해 질량이 d만큼 변했다는 것이다.
Given the frequent and common modification list in advance, the function can be adjusted. In this work, we considered pf(d, aa) as a constant C for all of the modifications
특이한 케이스를 피하기 위해서 T는 항상 길이가 0인 special tag t0, tv를 포함한다고 가정한다.
t0: 펩타이드 P의 position zero(시작 위치를 의미하는 듯)
tv: n (태그의 총 개수이다)
The overall best candicate score is obtained from M[n][tv][0].
MODa에서는 scoring을 위해서 스펙트럼 S를 PRM(Prefix Residue Mass) spectrum으로 변환한다. 먼저, S는 100 m/z 단위의 창으로 분리되고 그 안에서 intensity가 높은 10의 peak만 남긴다. 이 peak들은 intensirt의 순위에 따라 가중치가 붙는다.
PRM node의 점수는 PRM에서 예상되는 이온 피크의 가중치 합계로 정의된다.
어떤 피크라도 2가 피크로 가정할 경우(이 경우는 1Da 위치에 parent peak이 있는지 여부에 따라 결정된다.)
MODa Workflow
- 각각의 MS2 spectru을 통해 나온 sequence tag를 이용해서 candidata peptide들을 DB에서 뽑아온다.
- dp 알고리즘을 이용해서 매치된 시퀀스 태그 사이에서의 modification을 identify한다.
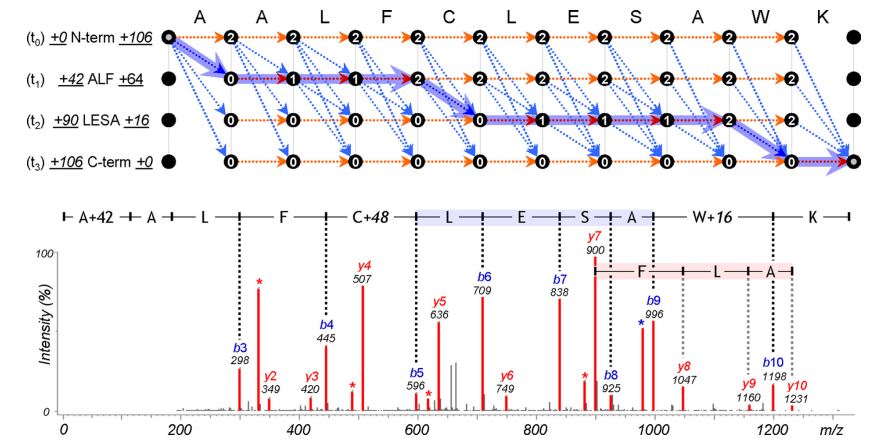
Fig 6
지금 위에 보이는 그림은 triply modified peptide이다. 즉 3번 modification이 일어난 펩타이드임
3번 modification이 일어났다면, tag는 t0, t1, t2, t3로 4개가 된다.
t0, t3는 각각 N-terminal과 C-terminal 쪽에 있는, 길이가 0인 tag들이고, 각각의 Modification 사이사이에 tag가 하나씩 껴있기 때문에 2개의 태그가 더해져서 총 4개의 태그가 존재한다.
여기서 C와 W에 modification이 발생했기 때문에 t1 = ALF, t2 = LESA가 된다.
각각의 row에서 node는 matched tag에 대한 위치에 따라 다음과 같이 분류된다.
- 0: tag의 시작 지점을 포함한 tag의 전 부분
- 1: tag의 inside
- 2: tag의 끝 지점을 포함한 tag의 뒷 부분
이것들은 노드 사이의 점프를 막기 위해서 사용된다.
amino acid jump는 같은 row에 대해서만 가능하기 때문
modification jump는 아래 규칙이 적용된다.
- 0으로 라벨링된 node에서의 점프는 불가능
- 1, 2 labeled node에서의 점프는 0, 1 labeled node로 가능
- 2번의 연속된 점프는 그 사이에 적어도 한개 이상의 tag가 있다면 가능(?) (그 태그 안에서 적어도 한번의 amino acid jump가 필요하다) -> 확실 X -> These rules ensure that dynamic programming must use at least out tag out of matched tags
matched tag 말고 적어도 하나의 태그를 이용을 해야함
하지만 모든 matched tag를 이용할 필요는 없음
Under these constraints, dashed arrows of Fig. 6 represent all possible jumps between nodes.
Here, the spectral alignment problem is finding the highest scoring path, where the score of a path is the sum of node scores on the path and each node's score is calculated based on the intensity of the corresponding peak in the experimental spectrum.

글 잘 봤습니다.