GENNIUS
Introduction
기존 고속 처리 실험 기술이 약물 개발 과정을 가속하기 위해 개발되었지만, in vitro 및 in vivo 실험은 여전히 시간이 오래 걸리고 비용이 많이 든다. 이러한 한계를 해결하기 위해 계산 기반 방법들이 등장하였으며, 이는 새로운 치료제를 시장에 출시하는 데 필요한 시간과 자원을 줄이는 데 유망한 도구로 여겨진다.
최근에는 대량의 데이터를 기반으로 이전에 발견되지 않았던 복잡한 분자 패턴을 식별할 수 있는 머신러닝 모델을 설계할 수 있게 되었다. 이들 중 일부는 계산 비용이 매우 높아, 약물 - 표적 상호작용을 더 잘 표현할 수 있는 대규모 네트워크를 사용하는 데 제한이 있으며, 실행 시간조차 보고되지 않는 경우도 많았다. 또한 대부분의 기술은 단백질과 약물이 어떻게 연결되어 있는지에 대한 전체적인 관점을 고려하지 않으며, 이는 새로운 관계를 발견하는 데 중요한 정보가 될 수 있다.
이러한 네트워크 구조를 모델링하기 위해, 최근에는 DTI 데이터를 그래프로 표현하는 방법이 제안되었다. 약물과 단백질은 노드로, 실험을 통해 확인된 상호작용은 엣지로 표현되는 이종 그래프로 모델링될 수 있다. 이 그래프는 질병과 같은 추가적인 노드나 단백질 유사도와 같은 엣지를 추가하여 확장될 수 있다. 이후 모델은 특정 약물이 단백질과 상호작용할 가능성을 예측하도록 학습된다.
이 논문은 SAGEConv 레이어를 기반으로 하고, 이후 신경망 분류기를 결합한 새로운 DTI 예측 모델 GENNIUS를 제안한다.
Materials and Methods
Model architecture
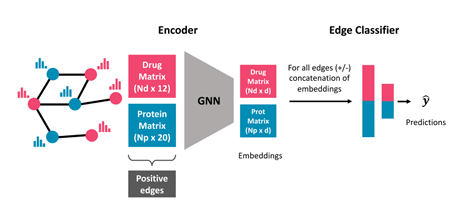
노드 임베딩을 생성하는 GNN 인코더, 약물과 단백질 노드 임베딩을 결합하여 엣지의 존재 여부를 학습하는 NN기반 분류기로 구성된다.
GNN에서는 그래프의 노드들은 이웃들과 메시지를 교환하여 자신의 특징 표현을 업데이트 하며, 이는 두 가지 기본 함수인 메시지 함수와 업데이트 함수로 이루어진다.
- : 레이어
- : 노드 에 대한 집계된 메시지 벡터
- : 노드 의 이웃 노드
- : 차원을 가지는 노드 임베딩
- : 노드 와 이웃노드 사이의 메시지를 정의
- : 노드 와 이웃노드 사이 엣지 정보
- : 노드 업데이트 함수, 노드의 이웃으로부터 집계된 메시지와 해당 노드 자신의 표현을 결합한다.
GENNIUS의 인코더는 네 개의 SAGEConv 레이어로 구성되어 있으며, 각 노드의 지역 이웃의 임베딩으로부터 정보를 집계함으로써 네트워크 구조를 보존하는 노드 임베딩()을 생성하는 역할을 한다. 따라서 번째 SAGEConv 레이어에서 노드 의 임베딩은 다음과 같이 주어진다.
- : 활성화 함수
- AGG : 집계 함수
- : 학습 가능한 가중치 행렬
초기 feature 차원은 서로 다르다. drug feature : , protein feature : . 이때 GNN에서는 이웃 노드의 정보를 가져와 내 임베딩을 업데이트 한다. 하지만 drug는 protein 정보만 받고 protein은 drug 정보만 받는다. 그래서 첫 번째 레이어가 특별하다
- drug 업데이트
입력 : protein feature ()
출력 : drug embedding ()
그래서 이때 필요한 행렬은 - protein 업데이트
입력 : drug featuer ()
출력 : protein embedding()
그래서 이때 필요한 행렬은
그러므로 서로 입력 차원이 다르므로 가중치 크기도 다르다
k > 1 레이어. 즉, 두 번째 레이어부터는 모든 노드가 같은 차원 로 임베딩되므로 입력도 . 출력도 이므로 가중치는 로 통일된다.
NN 기반 분류기는 두 개의 완전연결 레이어로 구성되며, 두 레이어 모두 ReLU 활성화 함수를 사용하고, 마지막 출력층은 sigmoid 활성화 함수를 갖는 하나의 뉴런으로 구성된다. 분류기의 입력은 약물과 단백질 임베딩을 연결한 차원의 벡터이며, 출력은 상호작용이 존재할 확률의 추정값이다.
Model configuration
모델은 Adam 옵티마이저와 learning_rate 0.01로 학습되었다. 출력층의 sigmoide와 binary cross entropy를 하나의 함수로 결합한 손실 함수를 사용하였다. 이러한 결합은 수치적 안정성을 위해 log-sum-exp 기법을 활용한다.
배치 크기 으로 나누어진 데이터셋이 주어졌을 때, 배치 내 샘플 에 대한 손실 은 다음과 같이 계산된다.
- : 샘플 의 정답 라벨
- : 해당 샘플이 양성 클래스에 속할 확률의 추정값
- : 활성화 함수 이전의 마지막 선형 레이어의 출력
최종 배치 손실 은 의 평균으로 계산된다.
노드 특징 간 잠재적 다중공선성을 해결하기 위해 인코더 단계에 0.2의 dropout이 사용된다.
모델은 검증 데이터 기반으로 계산되는 early stopping을 사용하며, 최소 40 epoch의 학습을 수행한다. 이는 특히 작은 데이터셋에서 최반 epoch에서 early stop이 발생하여 underfitting이 되는 것을 방지하기 위함이다.
Model training and evaluation
-
학습
- Adam optimizer + learning rate 0.01
- loss = binary cross entropy (sigmoid 포함)
- dropout 0.2 - 과적합 & feature 중복 문제 완화
- early stopping - 너무 빨리 학습 멈추는 문제 방지
-
데이터 처리
- 그래프를 70 : 10 : 20로 나눔
- positive : negative = 1 : 1
-
중요한 포인트
GNN은 일부 데이터로 학습
나머지 데이터는 "엣지 예측" 학습에 따로 사용
-
평가
AUROC
AUPRC
Node features
약물과 단백질의 성질이 서로 다르므로 약물 노드와 단백질 노드에 대해 매우 다른 종류와 차원의 특징을 선택한다.
단백질 노드 특징은 20차원 벡터로 인코딩되며, 이는 20가지 서로 다른 아미노산을 반영한다. 각 특징은 해당 노드와 연결된 단백질 서열에서 특정 아미노산의 비율을 나타낸다.
약물 노드 특징은 RDKit을 사용하여 SMILES로부터 계산된 잘 알려진 분자 descriptors로 선택된다. (LogP, 분자량, 수소 결합 수용체의 수, 수소 결합 공여자의 수, 헤테로 원자의 수, 회전 가능한 결합의 수, 위상학적 극성 표면적, 고리의 수 및 방향족 고리의 수, NH와 OH의 수, 질소와 산소 원자의 수, 분자 내 heavy atom의 수, 원자가 전자의 수)
일부 특징은 서로 겹치지만 dropout으로 문제를 완화하였고, 더 좋은 특징(pretraining embedding)도 시도했지만 성능이 거의 안 좋아졌다. 오히려 pretrained feature는 일부 노드에만 존재하고 데이터가 줄어들어 성능이 악화되었다.
즉, 약물은 화학 특징, 단백질은 아미노산 비율로 표현해서 GNN 입력으로 사용한다.
Related Work
| 종류 | 설명 |
|---|---|
| DTINet | 다양한 관계를 포함한 큰 그래프 + matrix completion |
| EEG-DTI | GCN으로 임베딩 - 내적으로 예측 |
| HyperAttentionDTI | SMILES + sequence - CNN + attention |
| MolTrans | transformer 기반 서브구조 학습 |
| MCL-DTI | 멀티모달 + attention |
Datasets
| 종류 | 설명 |
|---|---|
| DrugBank | 가장 대표적인 약물 - 타겟 데이터 |
| BioSNAP | 실제 시장 약물 기반 데이터 |
| BindingDB | 실험 기반 binding affinity 데이터 |
| Davis | kinase 중심 데이터 |
| Yamanishi | 4개 서브셋(E, GPCR, IC, NR) DTI 분야 표준 데이터셋 |
SMILES 없는 drug는 제거하고 sequence 없는 protein은 제거하였으며, 대부분 데이터셋은 positive만 있어 negative edge는 랜덤 샘플링하여 positive:negative = 1:1로 하였다. 데이터는 서로 다른 시기에 만들어졌으므로 같은 DTI가 여러 데이터셋에 중복 존재할 수 있다.
Dataset configuration for interring unknown positives
한 데이터셋에서는 negative인데 다른 데이터셋에서는 positive인 경우를 활용하여 모델이 진짜 숨겨진 interaction을 맞추는지 평가한다.
Data leakage prevention during evaluation on unseen datasets
train과 test에 같은 edge가 있으면 모델이 이미 답을 아는 상태이므로 train과 test에 같은 positive edge를 제거하여 이를 해결한다. 하지만 negative edge는 유지한. 데이터에 없다고 해서 진짜 없는 건 아니기 때문이다.
Result
GENNIUS outperforms state-of-the-art methods
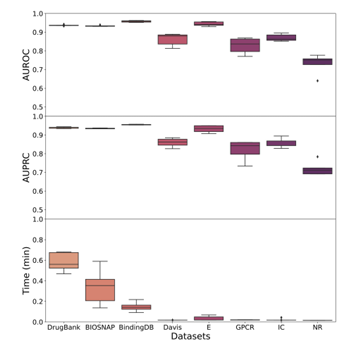
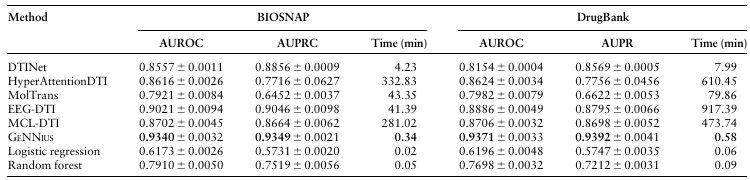
GENNIUS는 모든 데이터셋에서 높은 성능을 보였으며 기존 방법들 보다 속도가 압도적으로 빨랐다. EEG-DTI와 성능은 비슷하지만 속도는 1000배 이상 빨랐고, 단순모델보다도 크게 성능이 향상되었음을 보여준다.
GENNIUS prediction capabilities
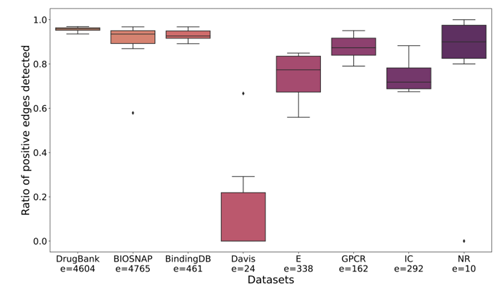
다른 데이터셋에서는 positive인데 여기서는 negative인 edge 테스트
큰 데이터셋에서는 80 ~ 90% 이상 맞추었고, 작은 데이터셋에서는 70% 정도를 맞추었음
GENNIUS generalization capabilities
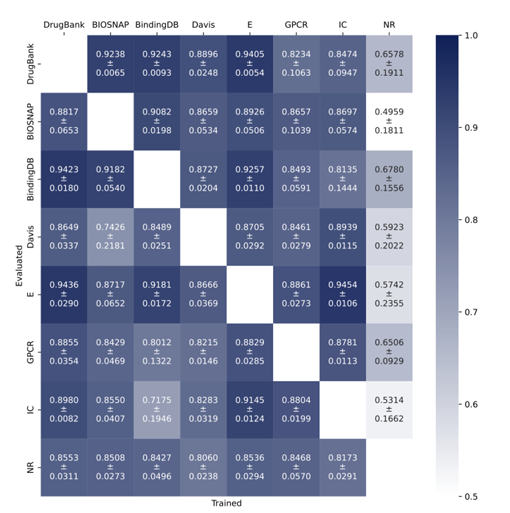
한 데이터셋에서 학습하고 다른 데이터셋에서 테스트 하였을 때, 큰 데이터셋으로 학습하면 다른 데이터셋에서도 성능이 좋았으며, 작은 데이터셋으로 학습하면 일반화에 실패하였다. 즉, 큰 그래프로 학습하면 작은 그래프에도 잘 적용됨을 보여준다.
GENNIUS encoder preserves biological information
GNN 임베딩이 의미 있는지를 t-SNE로 분석하였다
- Edge 수준에서는 약물/단백질 정보가 유지되었다
- Node 수준에서는 원래 단백질은 잘 구분되지 않았지만, GNN 이후에 protein family가 구분되었다. 즉, GNN이 정보를 diffusion함을 보여준다.
