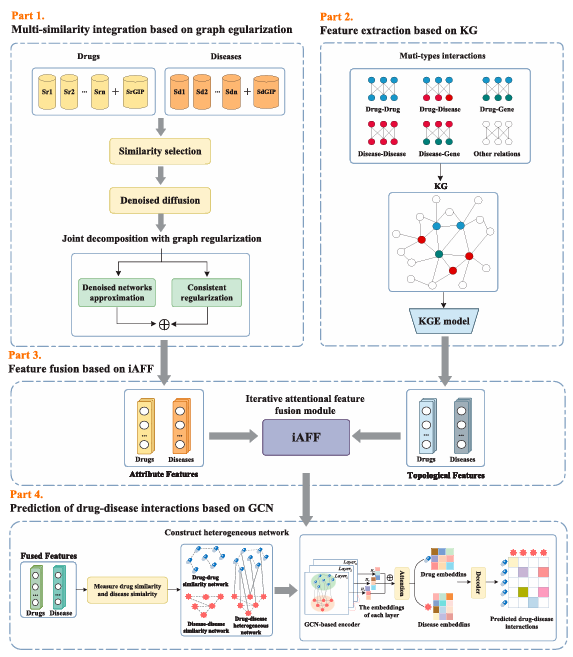
KGRDR
Introduction
기존 신약 개발 과정이 지나치게 비용이 크고 시간이 오래 걸리며 성공률이 낮았다(평균 12년 & 20억 달러 이상 비용 소요). 이러한 배경에서 등장한 것이 drug repositioning으로, 이미 승인된 약물에서 새로운 치료 효과를 찾는 방법이다. 이 접근법은 비용과 위험을 줄이고 개발 시간을 단축할 수 있다.
기존 계산적 drug repositioning 방법들은 주로 약물과 질병 간의 similarity 정보를 이용해 상호작용을 예측해왔다. 하지만 단일 종류의 similarity만 사용하여 정보가 부족하고 데이터가 희소하며 일반화 성능이 떨어지는 문제가 있었다.
이를 개선하기 위해 여러 종류의 similarity를 통합하는 방법들이 제안되었는데, 선형 방식은 단순하지만 복잡한 관계를 반영하지 못하고, 비선형 방식은 구조적 관계를 반영할 수 있지만 계산 비용이 크고 노이즈와 중복 정보 문제가 존재한다. 또한 matrix decomposition 기반 방법들도 제안되었지만 여전히 완전한 해결책은 아니었다.
또한, 약물과 질병뿐만 아니라 유전자, 경로 등 다양한 생물학적 엔티티 간 관계를 활용하기 위해 지식 그래프를 활용하여 풍부한 특징을 학습하려는 연구가 증가하고 있다. 이러한 접근은 약물과 질병 간의 구조적 및 맥락적 정보를 반영하여 예측 성능을 향상시키는 데 기여한다.
Materials and methods
Notations and brief reviews of KGRDR
- : 전체 유사도 집합
- : 약물 쌍 유사도 행렬
- : 약물 노드 개수
- : 질병 쌍 유사도 행렬
- : 질병 노드 개수
- : 번째 network에서 node 와 사이의 유사도 점수
- : 선택된 유사도 집합 (중복 제거)
- : 노이즈 제거된 유사도
- : 공통 속성 특징
- : 구조적 특징
- : 융합 특징
Multi-similarity integration based on graph regularization
다중 유사도 네트워크 내의 노이즈와 중복 정보가 모델의 예측 성능에 큰 영향을 미친다는 점을 고려해서, 약물과 질병의 다중 유사도 정보를 정제하고 통합하기 위해 그래프 정규화 기반 통합 방법을 사용한다. denoised diffusion model 모듈은 여러 유사도 네트워크의 노이즈를 제거하는 데 사용되며, joint decomposition 모듈은 정제된 네트워크들을 융합하는 역할을 한다.
Drug and disease similarity data
약물과 질병 간의 pairwise 유사도 데이터를 SND 데이터셋과 SMFDD_S 데이터셋으로부터 가져왔다.
- SND
- 약물 유사도 10개, 질병 유사도 14개, 약물-질병 상호작용 포함 - SCMFDD_S
- 약물 유사도 5개, 질병 유사도 1개, 약물-질병 상호작용 포함
모든 유사도 값은 0 ~ 1을 가진다.
또한, 기존 데이터 외에 Gaussian interaction profile kernel similarity를 추가로 계산한다.
- 약물 벡터
- 질병 벡터
이 두 벡터를 기반으로 약물-약물 / 질병-질병 유사도를 계산한다. (가까우면 유사도는 1에 가깝고 멀면 0에 가깝다)
Similarity selection
약물 및 질병 관련 유사도 행렬들은 품질, 정보량, 그리고 상관성이 서로 크게 다르다. 이러한 데이터의 불일치성과 중복성은 노이즈를 유발할 수 있다. 정보량이 높고 중복이 적은 약물 및 질병 유사도 부분 집합을 통해 효과적인 방법을 사용한다.
이 휴리스틱 유사도 선택 과정은 네 단계로 구성된다.
1. 각 유사도 행렬의 평균 엔트로피 계산
2. 평균 엔트로피 값을 기준으로 행렬들을 순위화
3. 서로 다른 데이터 소스에서 생성된 유사도 행렬들 간의 유사도를 계산
4. 중복되는 유사도 행렬을 제거
계산된 프로파일 유사도와 SND 및 SCMFDDS 데이터셋에서 얻은 유사도들을 이용하여 유사도 선택을 수행할 수 있다. 이 선택 과정은 정보량이 풍부하면서도 중복이 최소화된 유사도 행렬들의 부분 집합 ${ S^{(v)} }{v=1}^{V}$ 를 추출하며, 여기서 V는 선택된 유사도 행렬의 개수를 의미한다.
Diffusion to denoise matrices
선택된 유사도 행렬에도 여전히 일부 노이즈가 존재할 수 있으므로, denoised diffusion 방법을 적용하여, 유사도 선택 단계에서 얻은 유사도 행렬 의 노이즈를 추가적으로 감소시킨다.
- : v 번째 노이즈 제거된 유사도 행렬
- : 의 고유벡터들로 이루어진 행렬
- : 의 고유값으로 이루어진 대각 행렬
- : 단위 행렬
Joint decomposition with graph regularization
노이즈가 제거된 를 공통 특징 벡터 로 나타내기 위해, 그래프 정규화 기반 통합 방법을 사용한다. 이는 joint deomposition과 그래프 정규화를 결합하여, 노이즈 제거된 유사도 행렬 부분 집합으로부터 공통 속성 특징 표현 를 학습한다.
Joint decomposition은 여러 네트워크를 통합적으로 분석하고, 네트워크 간의 불일치를 포착하며, 네트워크들 간의 상관관계를 탐색할 수 있다.
- 공통 특징 행렬 : 네트워크 간의 공통 성분을 표현하는 데 사용
- 특이 특징 행렬 : 번째 네트워크의 고유한 성분을 표현하는 데 사용
각 노이즈 제거된 행렬 는 재구성 행렬 로 근사되며(로 원래 유사도 복원하기), 로 정의된다. 와 간의 근사 오차는 KL divergence로 측정되며 다음과 같이 표현된다.
- : 의 열 벡터로서 v 번째 네트워크에서 노드 의 공통 특징 벡터
- : 의 열 벡터로서 v 번째 네트워크에서 노드 의 특이 특징 벡터
또한 공통 특징 가 로부터 정제된 구조 정보를 정확히 반영하도록 하기 위해, 그래프 라플라시안 정규화 항 가 에 도입된다. 구체적으로 모든 노드 쌍 간의 일관성은 다음과 같이 정의된다.
- : 의 대각 차수 행렬
- : 그래프 라플라시안 행렬
- : 행렬 트레이스
최종 목적 함수 식은 다음과 같이 정의 된다.
여기서 은 정규화 파라미터이며, 는 모든 유사도 행렬에서 공유되는 공통 속성 특징 표현으로 간주된다. 특징 표현 의 차원은 다중 유사도 네트워크의 통합 능력과 이후 예측 작업의 성능에 직접적인 영향을 미친다.
여러 개의 유사도 행렬은 서로 다른 데이터 소스에서 생성되기 때문에 서로 값이 다르고, 일부는 노이즈나 중복 정보를 포함하고 있다. 따라서 이를 그대로 사용할 경우 모델의 성능이 저하될 수 있으므로, 여러 유사도 행렬로부터 신뢰할 수 있는 핵심 정보만 추출할 필요가 있다.
또한 각 유사도는 서로 다른 측면의 부분적인 정보만을 담고 있다. 예를 들어 화학 구조 유사도는 약물의 형태적 특성만 반영하고, 유전자 기반 유사도는 생물학적 기능을, 네트워크 기반 유사도는 관계 정보를 반영한다. 따라서 하나의 유사도만으로는 전체적인 정보를 충분히 표현할 수 없으며, 여러 유사도를 통합해야 보다 완전한 표현이 가능하다.
하지만 단순히 여러 유사도를 결합하면 노이즈까지 함께 포함되기 때문에, 공통적으로 나타나는 패턴만을 추출하여 하나의 공통 특징 표현 𝑋로 만드는 과정이 필요하다.
한편, 유사도 행렬은 단순한 수치 데이터가 아니라 노드 간 관계를 나타내는 그래프 구조이므로, 이러한 구조적 정보가 유지되어야 한다. 이를 위해 그래프 정규화를 통해 유사한 노드들이 유사한 특징을 갖도록 한다.
또한 단순히 특징을 압축하면 중요한 정보가 손실될 수 있기 때문에, 원래의 유사도 행렬을 다시 복원할 수 있도록 학습(reconstruction)을 수행한다. 이를 통해 정보 손실을 최소화하면서도 의미 있는 공통 표현을 학습할 수 있다
Feature extraction based on KG
약물과 질병의 위상적 특징을 학습하기 위해 Drug Repurposing Knowledge Graph를 사용하였다. DRKG는 약물 재창출을 위해 특별히 설계된 지식 그래프로, 약물, 질병, 유전자 등의 엔티티와 이들 간의 관계를 포함하고 있다. 지식 그래프 임베딩은 이러한 엔티티 관계를 저차원 벡터 공간으로 매핑하여, 구조적 정보와 의미적 정보를 모두 보존한다. 여기서는 임베딩 표현을 학습하기 위해 CompLEx 지식 그래프 임베딩 방법을 적용한다.
Knowledge graph construction
DRKG는 13가지 엔티티 유형에 속하는 97,238개의 엔티티와 107가지 엣지 유형에 속하는 5,874,261개의 트리플을 포함하고 있다. 지식 그래프에 포함된 엔티티와 관계의 유형은 다음과 같다.
- 엔티티 : 질병, 약물, 유전자, 화합물 등
- 관계 : 약물-표적, 유전자-질병, 약물-질병 등
DRKG는 엔티티-관계-엔티티 형태의 트리플로 구성된다. (A, compound:Disease, B)라는 트리플은 약물 A가 질병 B와 상호작용함을 의미한다.
지식 그래프에서 엔티티는 노드로, 관계는 주체 엔티티 노드에서 객체 엔티티 노드로 향하는 엣지로 표현된다.
또한, DRKG에서 벤치마크 데이터셋에 존재하지 않는 약물-질병 관계 트리플을 모두 제거하고, 해당 데이터셋에 존재하는 새로운 약물-질병 관계 트리플을 DRKG에 추가하였다.
Knowledge graph embedding
ComplEx는 엔티티와 관계를 복소수 공간에 임베딩하여 모델링하며, 이를 통해 트리플(약물 - 질병 - 관계) 간의 비대칭 관계를 포착하면서 벡터 표현을 유지할 수 있어, 약물-질병 상호작용 예측 문제를 보다 효과적으로 처리할 수 있다.
약물 질병 (맞음), 질병 약물 (틀림)
일반 벡터는 구분이 어려우므로 복소수를 통해서 방향성을 표현한다.
-
Embedding Initialization
데이터셋에는 개 약물과 개의 질병이 포함되어 있다.
각 약물 에 대해, 복소수 벡터 를 학습한다.
질병 에 대해서도 복소수 벡터 를 학습한다.
약물-질병 상호작용 관계 에 대해서는 복소수 벡터 를 학습한다.
-
Predicting drug-disease interaction scores
학습된 약물과 질병의 복소수 임베딩 벡터 간의 상호작용 강도를 측정하기 위해 scoring function을 사용하며, 이를 통해 잠재적인 약물-질병 상호작용을 예측한다.- $\Theta$ : 모델 파라미터 - $w_r$ : 관계의 임베딩 벡터 - $e_s$ : 주체 벡터 - $e_o$ : 객체의 켤레 벡터 - $Re(x)$ : x의 실수부 - $Im(x)$ : x의 허수부복소수 공간에서 이므로 는 대칭적이고, 는 비대칭적이다.
트리플 가 지식 그래프에 존재할 확률 식은 다음과 같다 -
Model training and optimization
적절한 약물 및 질병 임베딩을 학습하기 위해, ComplEx 모델은 네거티브 샘플링과 손실 함수를 통해 임베딩을 최적화한다. -
Extracting features
학습된 모델을 사용하여, 약물과 질병 엔티티의 임베딩 벡터를 추출하며, 이를 전역적인 위상 구조 특징 표현 로 표현한다.
Feature fusion based on iAFF
그래프 정규화 모듈을 통해 얻어진 와 지식 그래프 임베딩으로부터 학습된 위상적 특징 를 융합하기 위해 iAFF 방법을 사용하였다.
iAFF는 특징 표현을 반복적으로 업데이트하며, MS-CAM(Multiscale Channel Attention Module)을 사용하여 각 채널에서 서로 다른 스케일의 특징 정보를 학습하고 각 채널 특징의 어텐션 가중치를 계산한다. 이러한 어텐션 가중치는 원래 특징에 원소별로 곱해져 채널 수준의 특징 강화가 이루어진다.
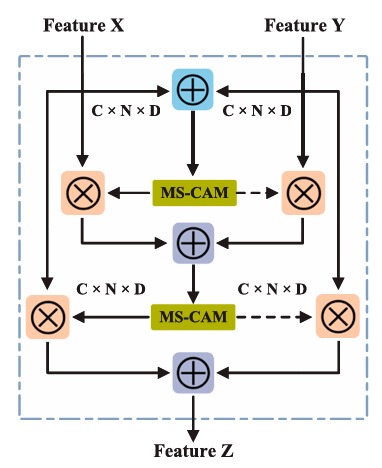
입력 특징 표현 와 는 CNN을 통해 추출되어 개의 채널과 크기의 특징 맵을 갖는 중간 특징으로 변환된다.
- 입력 특징 와 에 대해 초기 특징 융합
- 입력 특징 맵에 대해 보다 포괄적인 인식을 달성하기 위해 MS-CAM 모듈을 사용하여 의 어텐션 가중치를 계산하고, 특징 와 에 서로 다른 가중치를 부여한다.
- 어텐션으로 융합된 특징 는 MS-CAM 모듈에 반복적으로 입력되어 새로운 어텐션 가중치를 계산하며 특징 와 에 서로 다른 가중치를 다시 부여한다.
이러한 두 단계 계산 방식이 iAFF이다. 최종적으로 반복적 어텐션 융합 특징 는 다음과 같다.
Prediction of drug-disease interactions based on GCN
특징 융합을 통해 얻은 약물과 질병의 특징 벡터와 기존에 알려진 약물-질병 상호작용을 함께 그래프 합성곱 신경망에 입력하여 후보 약물-질병 상호작용을 예측한다.
Construction of the drug-disease heterogeneous network
융합된 약물 및 질병 특징 행렬을 이용하여, 유클리드 거리 기반으로 약물 유사도 행렬 과 질병 유사도 행렬 를 계산한다. 이후 약물 유사도 행렬 , 질병 유사도 행렬 , 그리고 약물-질병 상호작용 행렬을 사용하여 약물-질병 이종 네트워크를 구성한다.
- : 약물-약물 유사도 행렬 ( 개의 약물을 갖는 그래프)
- : 약물 유사도 행렬 로 구성됨
약물 유사도 행렬 에서 약물 의 top-k KNN에 약물 가 포함될 경우, 로 설정하고, 그렇지 않으면 으로 설정한다.
- : 질병-질병 유사도 행렬 (개의 질병을 갖는 그래프)
- :질병 유사도 행렬 로 구성됨
질병 의 top-k KNN에 질병 가 포함될 경우 , 그렇지 않으면 0으로 설정
약물-질병 상호작용 행렬은 N개의 약물과 M개의 질병을 갖는 그래프 G로 표현되며, 인접 행렬 이다. 약물 와 질병 사이에 연관성이 있으면 = 1, 그렇지 않으면 으로 설정한다.
Feature extraction based on GCN
- 약물-약물 유사도 네트워크과 질병-질병 유사도 네트워크를 사용하여 intra-domain 임베딩을 추출
- 질병-약물 연관 네트워크를 이용하여 inter-domain을 추출: l번째 층에서의 약물 intra-domain 출력 특징
: 질병 intra-domain 출력 특징
그래프 합성곱 식은 다음과 같다
inter-domain 특징 추출 모듈은 약물과 질병 간 메시지 전달을 위해 bilinear aggregator와 전통적인 GCN aggregator로 구성된다. 약물 에 대한 inter-domain 특징은 다음과 같다
- 이 두 임베딩을 결합하여 약물과 질병의 최종 임베딩을 얻는다.여러 GCN 층을 거치면서 서로 다른 수준의 정보를 학습하게 되며, L층 이후 k 차원의 약물 및 질병 임베딩을 얻는다.
이후 각 GCN 층의 임베딩을 결합하기 위해 layer attention을 도입한다. 각 층의 중요도를 반영하여 최종 임베딩 식은 다음과 같이 계산한다.
Drug-disease interaction prediction
약물과 질병 간의 연관성을 재구성하기 위해, 디코더 는 다음과 같이 정의된다
- : 예측된 확률 행렬로 약물 와 질병 간의 연관성 점수를 의미한다
모델 파라미터는 가중 이진 교차 엔트로피 손실을 최소화하도록 학습되며 다음과 같이 정의 된다.
여기서 는 알려진 약물-질병 연관 쌍의 집합, 는 알려지지 않았거나 관측되지 않은 쌍의 집합이다.
