1. 데이터 준비
우리는 전 게시글에서 ERR458493 의 Saccharomyces cerevisiae 샘플을 정렬하였다.
정렬의 결과물 중 _ReadsPerGene.out.tab 파일은 각 유전자에 몇 개의 시퀀싱 Read가 Mapping 되었는지 그 Count를 정리한 데이터 인데, 해당 데이터로 DEG 분석을 진행해보고자 한다.
우선적으로, ERR458493 는 Wild Type(WT) 샘플이며, 추가적으로 1개의 WT 샘플과 2개의 snf2돌연변이(MU) 샘플을 준비해 총 4개의 RNA-seq 샘플을 대상으로 분석을 진행하였다.
- 분석 대상
- 대조군(WT) :
ERR458493,ERR459175 - 실험군(MU) :
ERR458506,ERR458515
- 대조군(WT) :
해당 파일들을 각각 4회 STAR 정렬하여 , 4개의 ...ReadsPerGene.out.tab 파일이 있다는 전제로 진행하겠다.
2. Gene Count Matrix 생성
STAR의 --quantMode GeneCounts 옵션은 각 샘플에 대해 독립적인 유전자 발현량(Read Count) 파일을 생성한다. DEG 분석을 수행하기 위해 이 4개의 데이터들을 하나의 통합 행렬로 병합하여야한다. 이 행렬은 유전자(행) X 샘플(열)을 구조로 가진다.
2-1. ReadsPerGene.out.tab 살펴보기
N_unmapped 44500 44500 44500
N_multimapping 111531 111531 111531
N_noFeature 35757 445692 474155
N_ambiguous 60129 3329 3260
YDL246C 0 0 0
YDL243C 28 14 14
YDR387C 59 27 32
YDL094C 2 3 2
YDR438W 27 15 12
YDR523C 2 1 1
...다음은 ERR458493_ReadsPerGene.out.tab의 내용인데, 최 상단 4줄은 통계에 대한 요약 행이고, 그 아래로 GeneID, Unstranded Count, Starnded(Forward) Count, Strand(Reverse) Count 를 열로 가지는 데이터가 있다.
2-2. Count Matrix 생성
import pandas as pd
import os
BASE_DIR = "./alignment_output"
samples = {
"WT_1": "ERR458493_ReadsPerGene.out.tab",
"WT_2": "ERR459175_ReadsPerGene.out.tab",
"MU_1": "ERR458506_ReadsPerGene.out.tab",
"MU_2": "ERR458515_ReadsPerGene.out.tab"
}
counts_list = []
print("Create Matrix ...")
for sample , filename in samples.items():
file_path = os.path.join(BASE_DIR, filename)
if not os.path.exists(file_path):
print(f"ERROR: {file_path} not found")
continue
# 상단 4줄은 통계 요약이니 제외
df = pd.read_csv(file_path, sep='\t', header=None, skiprows=4)
# GeneID, Count만 추출 후 rename
df = df.iloc[:, [0,1]]
df.columns = ['GeneID', sample]
df.set_index('GeneID', inplace=True)
counts_list.append(df)
print(f"-{sample} load complete.")
if counts_list :
count_matrix = pd.concat(counts_list, axis = 1)
count_matrix.to_csv("count_matrix.csv")
print("COMPLETE : save [count_matrix.csv]")
print(count_matrix.head())
else :
print("ERROR : data not founds")해당 코드의 결과로 4개의 카운트가 하나로 병합된 count_matrix.csv가 생성된다.
GeneID,WT_1,WT_2,MU_1,MU_2
YDL246C,0,0,0,0
YDL243C,28,31,52,33
YDR387C,59,56,131,103
YDL094C,2,0,6,2
YDR438W,27,23,67,46
YDR523C,2,4,10,3
YDR542W,1,0,0,1
YDR492W,57,70,159,64
YDR018C,5,6,39,31
...이제, 이 파일을 가지고 DEG 분석을 실행 할 수 있다.\
3. 차등 발현 유전자 분석
3.1 Workflow
우리는 DEG분석을 위해 PyDESeq2 를 사용할 것인데, 이는 RNA-seqCount 데이터의 특성을 고려하여 통계적으로 유의미한 DEG를 식별하는 표준 도구이다.
정규화 - 분산 추정 - 가설 검정 - 다중 검정 보정 의 과정을 거치며 해당 내용은 추후에 게시글을 업로드 하겠다.
3.2 in Python code
import pandas as pd
import numpy as np
import matplotlib.pyplot as plt
from pydeseq2.dds import DeseqDataSet
from pydeseq2.ds import DeseqStats
print("Load data...")
counts_df = pd.read_csv("count_matrix.csv", index_col = 0)
# PyDESeq2 는 sample X gene 형태를 요구한다.
counts_df = counts_df.T
counts_df = counts_df.fillna(0)
counts_df = counts_df.astype(int)
# 샘플 합계가 10 미만인 노이즈 유전자 제거
counts_df = counts_df.loc[:, counts_df.sum(axis=0) >= 10]
print(f"-> analsis target : {counts_df.shape[0]} sample, {counts_df.shape[1]} gene")
# Create Metadata
metadata = pd.DataFrame(index=counts_df.index)
metadata['condition'] = ['WT' if 'WT' in sample else 'MU' for sample in metadata.index]
print("-> Metadata:")
print(metadata)
# DESeq2
print("analsis DESeq2...")
dds = DeseqDataSet(
counts=counts_df,
metadata=metadata,
design_factors='condition', # 기준 열
ref_level=["condition","WT"], # Reference is WT
n_cpus=4
)
dds.deseq2() # 정규화 및 분산 추정
# 통계분석
stat_res = DeseqStats(dds, contrast=["condition","MU","WT"], n_cpus=4)
stat_res.summary()
res = stat_res.results_df
res.to_csv("DESeq2_results.csv")
print("save to [DESeq2_results.csv]")
# 시각화
print("\n3. 볼케이노 플롯 생성 중...")
# 시각화를 위한 데이터 준비 (p-value 없는 것 제거)
plot_data = res.dropna(subset=['padj', 'log2FoldChange']).copy()
# 색상 기준 설정
# Red: 유의미하게 증가 (Up) / Blue: 유의미하게 감소 (Down) / Grey: 의미 없음
conditions = [
(plot_data['log2FoldChange'] > 1) & (plot_data['padj'] < 0.05),
(plot_data['log2FoldChange'] < -1) & (plot_data['padj'] < 0.05)
]
choices = ['red', 'blue']
plot_data['color'] = np.select(conditions, choices, default='lightgrey')
# 그래프 그리기
plt.figure(figsize=(10, 8))
# 모든 점 찍기
plt.scatter(
plot_data['log2FoldChange'],
-np.log10(plot_data['padj']),
c=plot_data['color'],
alpha=0.6,
s=15
)
# 기준선 그리기
plt.axvline(x=1, color='black', linestyle='--', linewidth=0.8) # 2배 증가
plt.axvline(x=-1, color='black', linestyle='--', linewidth=0.8) # 2배 감소
plt.axhline(y=-np.log10(0.05), color='black', linestyle='--', linewidth=0.8) # p-value 0.05
plt.title("Volcano Plot: Mutant vs WT (PyDESeq2)", fontsize=15)
plt.xlabel("log2 Fold Change", fontsize=12)
plt.ylabel("-log10 Adjusted P-value", fontsize=12)
top_genes = plot_data.sort_values('padj').head(5)
for gene_name, row in top_genes.iterrows():
plt.text(
row['log2FoldChange'],
-np.log10(row['padj']) + 0.2,
gene_name,
fontsize=9,
ha='center',
color='black',
fontweight='bold'
)
# 저장 및 출력
plt.tight_layout()
plt.savefig("Volcano_Plot_Python.png", dpi=300)
print("[완료] 그래프가 'Volcano_Plot_Python.png'에 저장되었습니다.")
plt.show()4. 결론
위 단계를 통해 우리는 DEG 결과인 DESeq2_results.csv와 Volcano_plot을 얻었다.
4.1 .csv
GeneID,baseMean,log2FoldChange,lfcSE,stat,pvalue,padj
YDL243C,35.23241178557316,-0.46323511752066077,0.4113790443156044,-1.1260542410256393,0.26014256106731215,0.5698109731025939
YDR387C,81.22576980096602,0.04681292104056832,0.2963043850078386,0.15798929549871463,0.8744652337332883,0.9598669278117801
...해당 파일에서 가장 중요한 열은 GeneID, log2FoldChange,padj 이다.
해당 파일에서 padj < 0.05 && |log2FoldChange| > 1 인 유전자를 필터링 하여 유의미한 최종 DEG 목록을 확인할 수 있다.
4.2 Volcano_plot
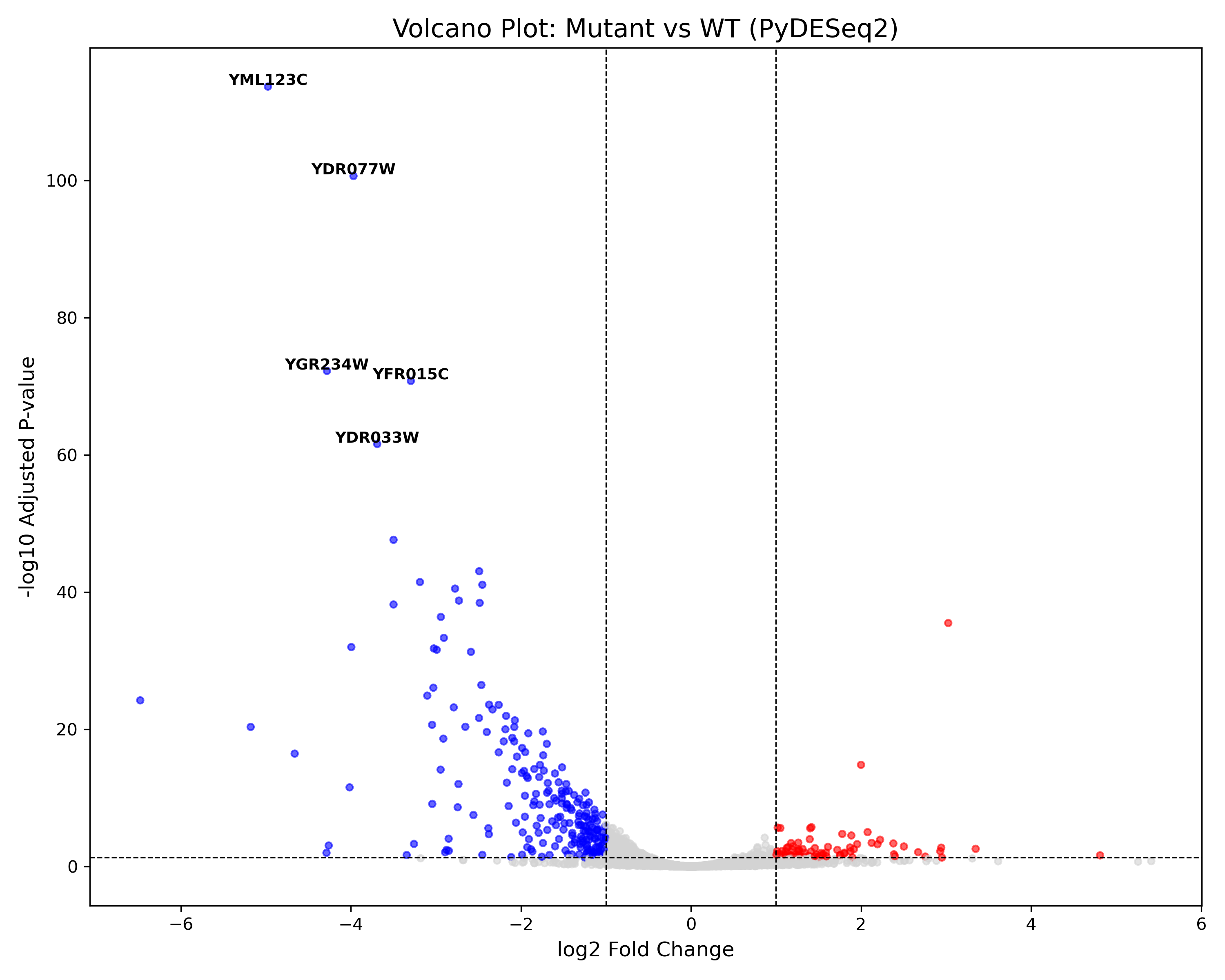
이 이미지에서 파란색 그룹은 DOWN그룹으로 유의미하게 발현이 감소한 그룹을 의미하고 빨간색 그룹은 UP 그룹으로 유의미하게 발현이 증가한 그룹을 의미한다.
이 최종 DEG 목록(UP 그룹, DOWN 그룹)을 유전자 기능 강화 분석 (GO Enrichment Analysis) 또는 경로 분석 (KEGG Pathway Analysis)에 사용하여 생물학적 의미를 해석하는 후속 연구로 이어질 수 있다.
