그동안 우리는 효모 유전정보를 활용하여 정렬하고 DEG를 해보았는데, 이는 bulk RNA-seq로 조직 전체 또는 수백만개의 세포의 평균을 분석하는 방법이다.
이제 더 나아가서 개별 세포 하나하나를 분석하는 Single-Cell RNA-seq(scRNA-seq) 를 해보고자 한다.
scRNA-seq를 위해 R에서는 Seurat 라이브러리를 활용하는데, 해당 홈페이지에서 튜토리얼과 10X Genomic에서 제공하는 2700개의 말초 혈액단백구 Peripheral Blood Mononuclear Cells(PBMC)의 데이터를 다운받을 수 있다.
1. library & data load
library(dplyr) # 데이터 집계 및 처리용
library(Seurat) # scRNAseq
library(patchwork) # 그래프
# 2700개의 Peripheral Blood MononuclearCells(PBMC) data Load
pbmc.data <- Read10X(data.dir= "/Users/juno/Documents/GitHub/seurat_prac/filtered_gene_bc_matrices/hg19")
# Seurat Object 지정
pbmc <- CreateSeuratObject(
counts = pbmc.data, # row data 지정
project = "pbmc3k", # 프로젝트 이름
min.cells = 3, #최소 3개 이상의 세포에서 발견된 유전자만 남김
min.features = 200 #유전자가 최소 200개 이상 검출된 세포만 남김
)dplyr: 데이터 가공Seurat: scRNA-seqpatchwork: 그래프 배치
그리고CreateSeuratObject를 활용해서 Seurat object를 생성하는데,min.cells와min.features를 이용하여 너무 적은 유전자만 가진 세포나, 너무 적은 세포에서만 발현된 유전자를 1차로 필터링 한다.
2. Quality Control
데이터의 노이즈를 필터링하여 분적의 정확도를 높인다. 여기서, 노이즈는 다음 경우들로 간주한다.
- 죽은 세포 : 미토콘드리아 유전자(
^MT-) 비율이 비정상적으로 높은 세포
- 싱글 셀 실험과정 중에 세포막이 깨지면(Lysis), 세포질 RNA는 유실되지만 미토콘드리아는 막으로 싸여있어 RNA가 보존되므로 죽은 세포 이거나 상태가 안좋은 세포의 경우 상대적으로 미토콘드리아 유전자 비율이 높게 잡힌다.
- Empty Droplets : 유전자 수가 너무 적은 경우
- Doublets(두 세포가 뭉친 경우) : 유전자 수가 비정상적으로 많은 경우
# 미토콘드리아 비율 계산
pbmc[["percent.mt"]] <- PercentageFeatureSet(pbmc, pattern = "^MT-")
# QC 지표 시각화
VlnPlot(pbmc, features = c("nFeature_RNA", "nCount_RNA", "percent.mt"), ncol = 3)
# 필터링 (유전자 수 200~2500 사이, 미토콘드리아 < 5%)
pbmc <- subset(pbmc, subset = nFeature_RNA > 200 & nFeature_RNA < 2500 & percent.mt < 5)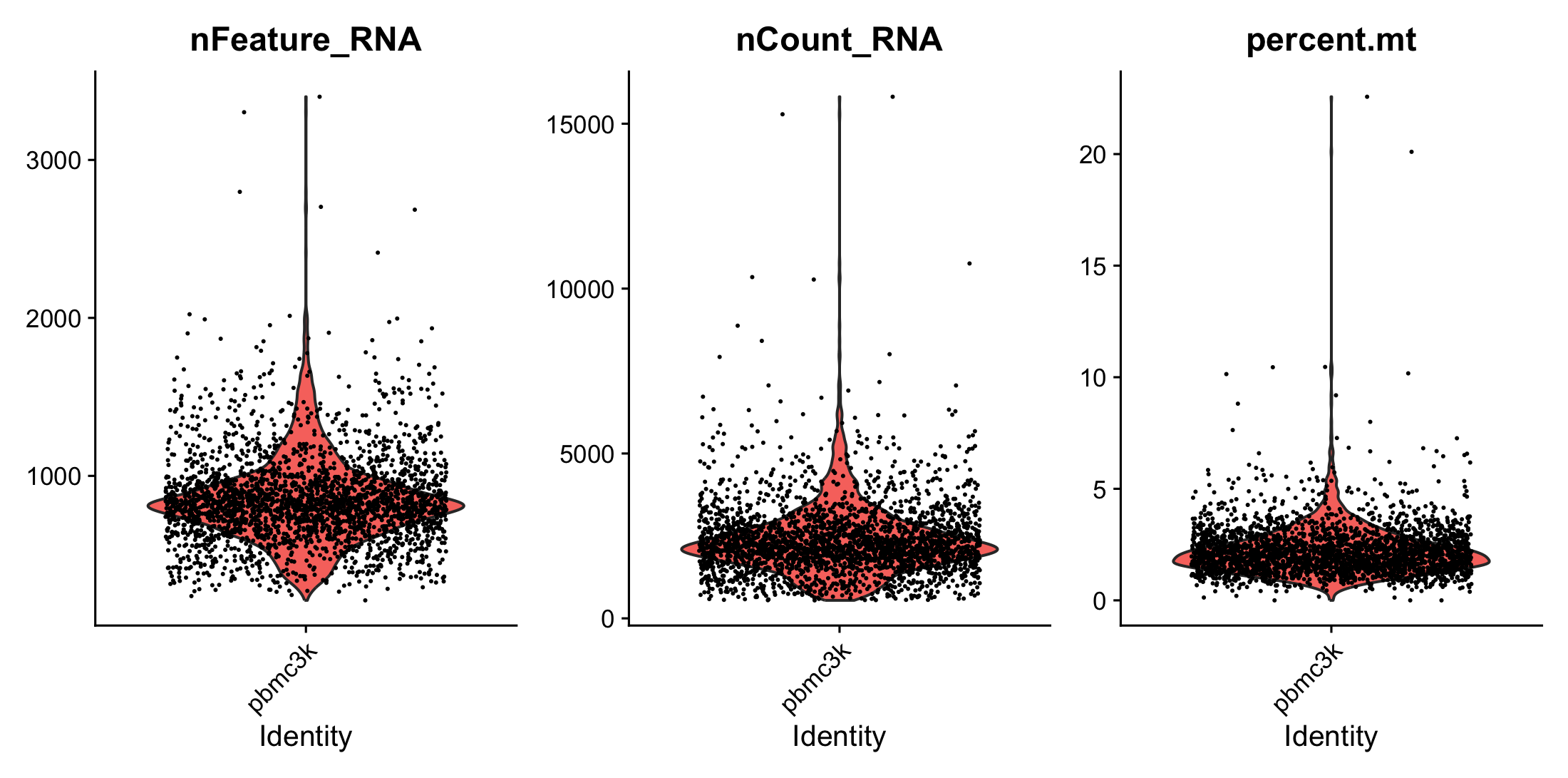
nFeature_RNA: 세포 하나에서 검출된 유전자의 가짓수nCount_RNA: 세포 하나에서 검출된 RNA 분자의 총 개수(UMI count)percent.mt: 전체 유전자 중 미토콘드리아 유전자가 차지하는 비율
3. Normalization & Feature Selection
세포마다 읽힌 RNA의 양이 다르기 때문에 정규화를 진행한 후 분석에 유의미한 Feature gene을 찾는다.
# Normalizing the data
pbmc <- NormalizeData(pbmc, normalization.method = "LogNormalize", scale.factor = 10000)
# 위의 파라미터들은 기본 값으로, pbmc <- NormalizeData(pbmc) 로 적용 가능
# highly variable features 식별
pbmc <- FindVariableFeatures(pbmc, selection.method = "vst", nfeatures = 2000)
top10 <- head(VariableFeatures(pbmc), 10)
plot1 <- VariableFeaturePlot(pbmc)
plot2 <- LabelPoints(plot = plot1, points = top10, repel = TRUE)
plot1 + plot2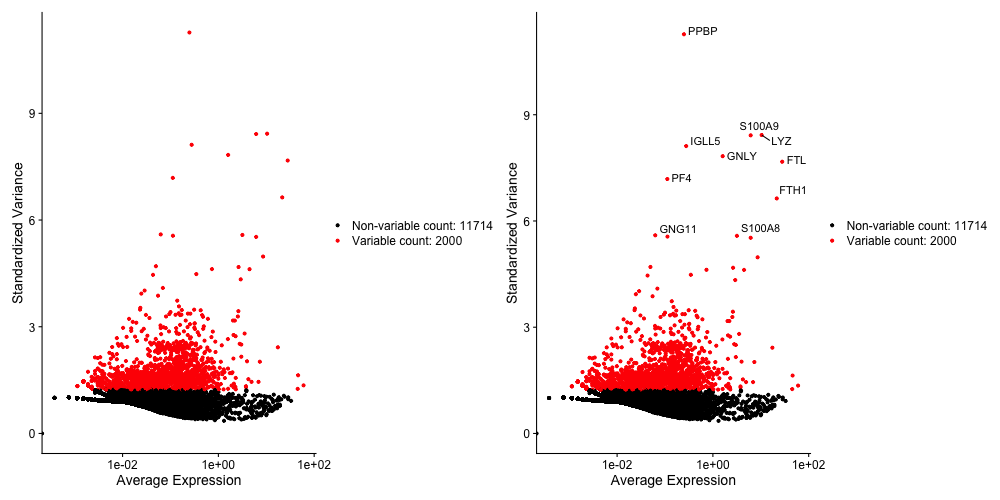
4. Scaling & PCA
데이터를 표준화(Scaling) 한 후, 주성분 분석(PCA)을 통해 데이터의 복잡도를 줄인다.
ScaleData(): 유전자 발현량의 평균을 0, 분산을 1로 조절한다. 발현량이 매우 높은 특징 유전자가 분석을 방해하는 것을 방지한다.RunPCA(): 수천 개의 유전자 정보를 50개 정도의 주성분으로 압축한다. 이를 통해 데이터의 주요 패턴을 요약 할 수 있다.
# PCA를 할수있도록 데이터의 차원을 축소한다.
# mean == 0, variance == 1
all.genes <- rownames(pbmc)
pbmc <- ScaleData(pbmc, features = all.genes)
# scaling이 끝난 데이터를 가지고 PCA (주성분 분석) 시작
pbmc <- RunPCA(pbmc, features = VariableFeatures(object = pbmc))5. Determine Dimensionality
PCA를 통해 압축된 주 성분 중 몇 개를 분석에 사용할 지 결정한다.
전통적으로 아래의 JackStraw 를 사용했지만 요즘은 ElbowPlot을 사용한다.
# 우리의 데이터는 여러 유전자의 발현도를 다양한 각도로 분석한 데이터를 가지고 있기 때문에
# 이것을 그래프로 표현하기 위해 차원을 축소해나가야 한다.
# JackStraw 를 활용한 data resampling (구버전)
pbmc <- JackStraw(pbmc, num.replicate = 100)
pbmc <- ScoreJackStraw(pbmc, dims = 1:20)
# 총 데이터에서 랜덤으로 1% 묶어 PCA를 돌려 차원을 축소하고, 그걸 이용해 null distribution을 만든다.
# 시간이 오래 걸린다 .
JackStrawPlot(pbmc, dims = 1:15)
ElbowPlot(pbmc)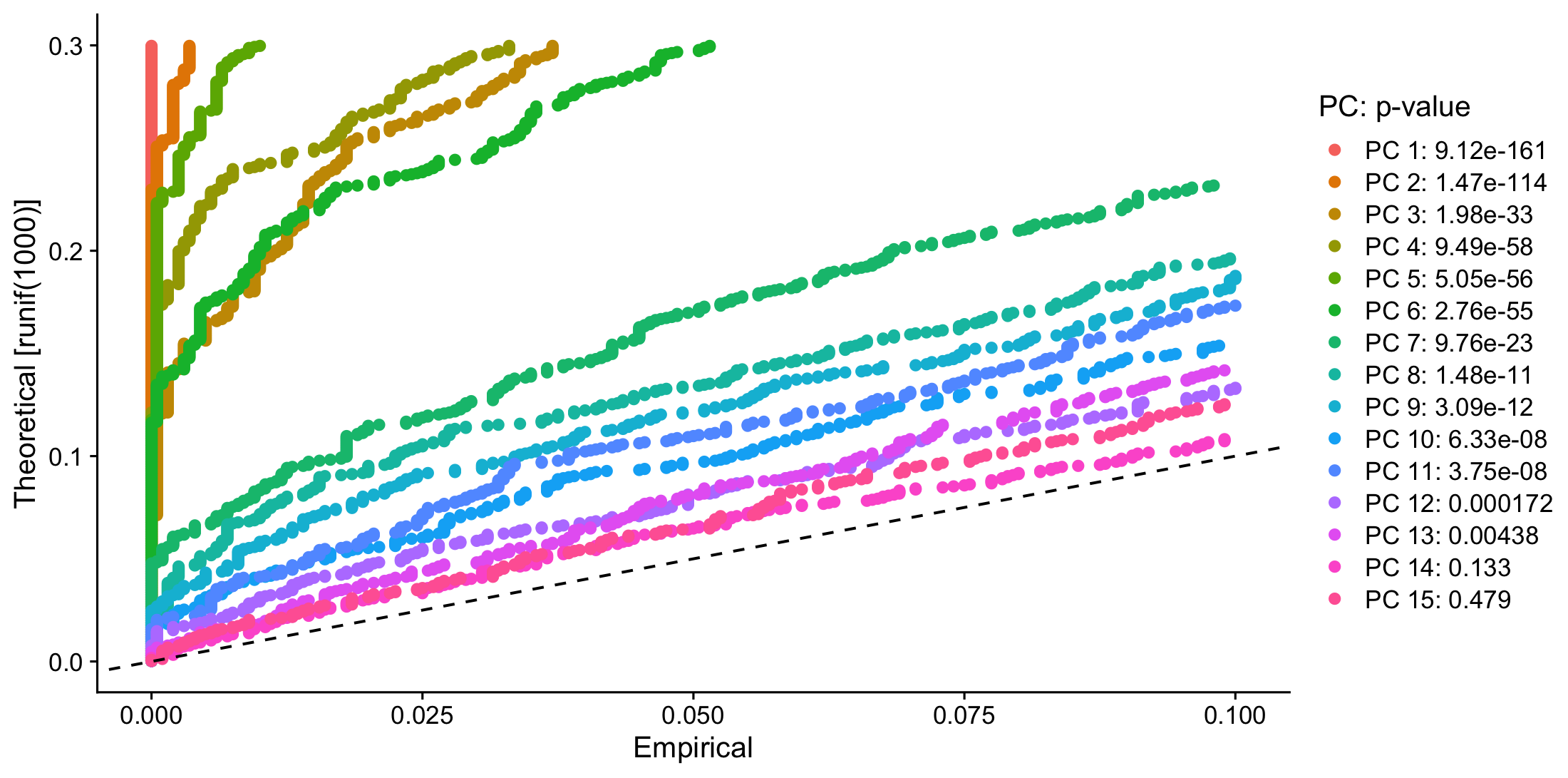
선형적인 추세를 가지는 PC가 7~12 사이 임을 알수있다. 그러므로 분석에 유의미한 PC는 처음부터 7~12 까지이다.
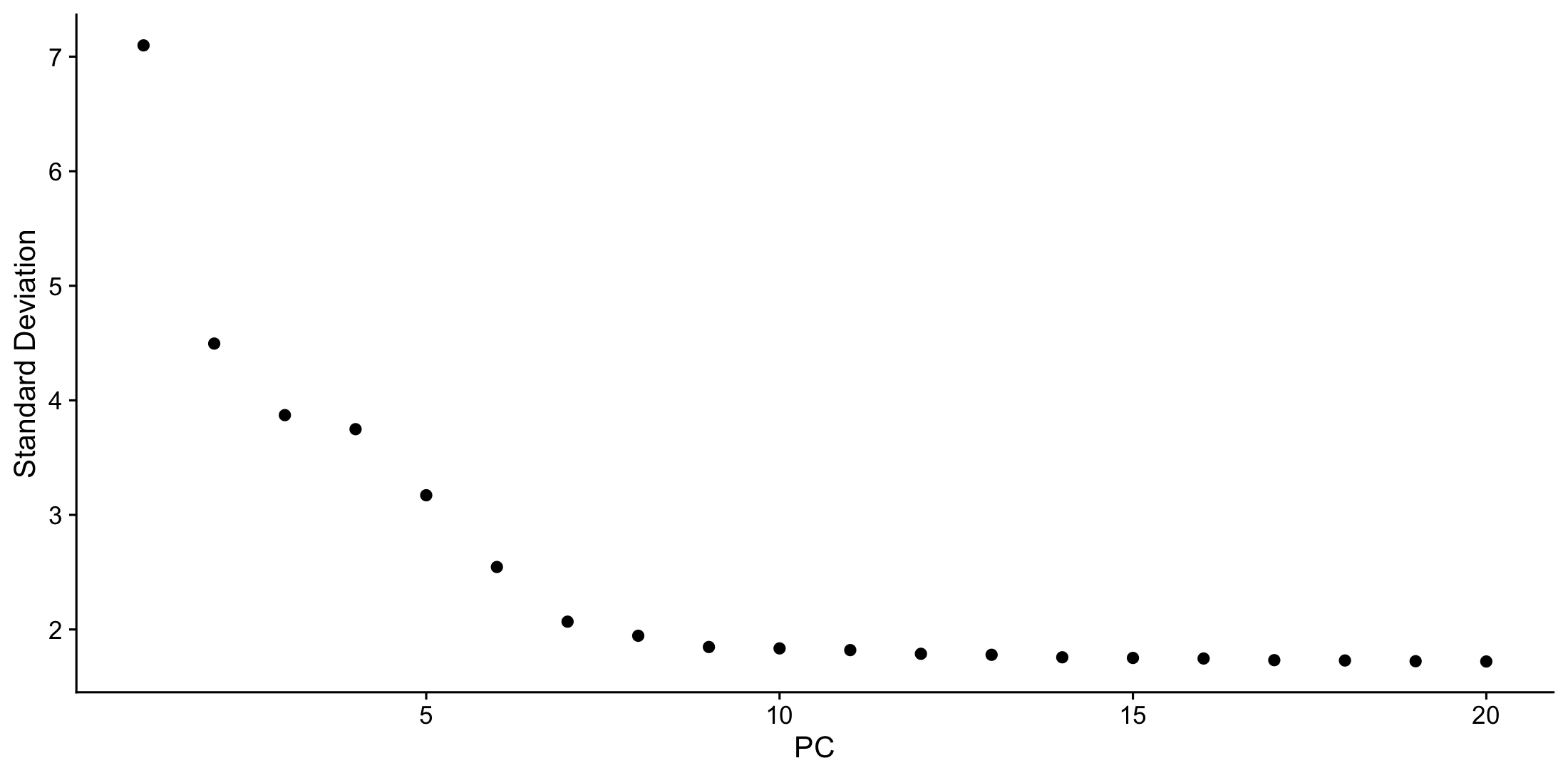
아래의 ElbowPlot을 보았을때, 감소 추세가 줄어드는 지점이 역시 PC 7~10 부근이므로, 역시 분석에 유의미한 PC는 0에서 부터 7~10까지이다.
6. Clustering
이제 우리는 PC를 10으로 기준점 삼아 유사한 유전자 발현 패턴을 가진 세포들끼리 Clustering을 할 것이다.
# 위에서 결정한 첫 10개의 PC만을 사용한다 .
pbmc <- FindNeighbors(pbmc, dims = 1:10) # PCA공간에서의 유클리드 거리 기반 KNN 그래프 구성
pbmc <- FindClusters(pbmc, resolution = 0.5) # resolution 은 세분화 정도를 지정.FindNeighbors: PCA공간에서 세포 간의 거리를 계산하여 그래프를 만든다.FindClusters: Louvain 알고리즘을 이용하여 세포들을 그룹화 한다.
7. 시각화 (UMAP / tSNE)
고차원 데이터를 우리가 볼 수 있는 2차원 평면에 투사한다.
- UMAP : 클러스터링 된 세포들이 2차원 상에서 끼리끼리 모여 있는지 시각적으로 확인한다. 가장 대중적인 시각화 방법이다.
# 비선형 차원 축소 (UMAP and tSNE)
# 클러스터링 결과를 시각화하고 데이터의 기본 구조 탐색
pbmc <- RunUMAP(pbmc, dims = 1:10)
DimPlot(pbmc, reduction = "umap")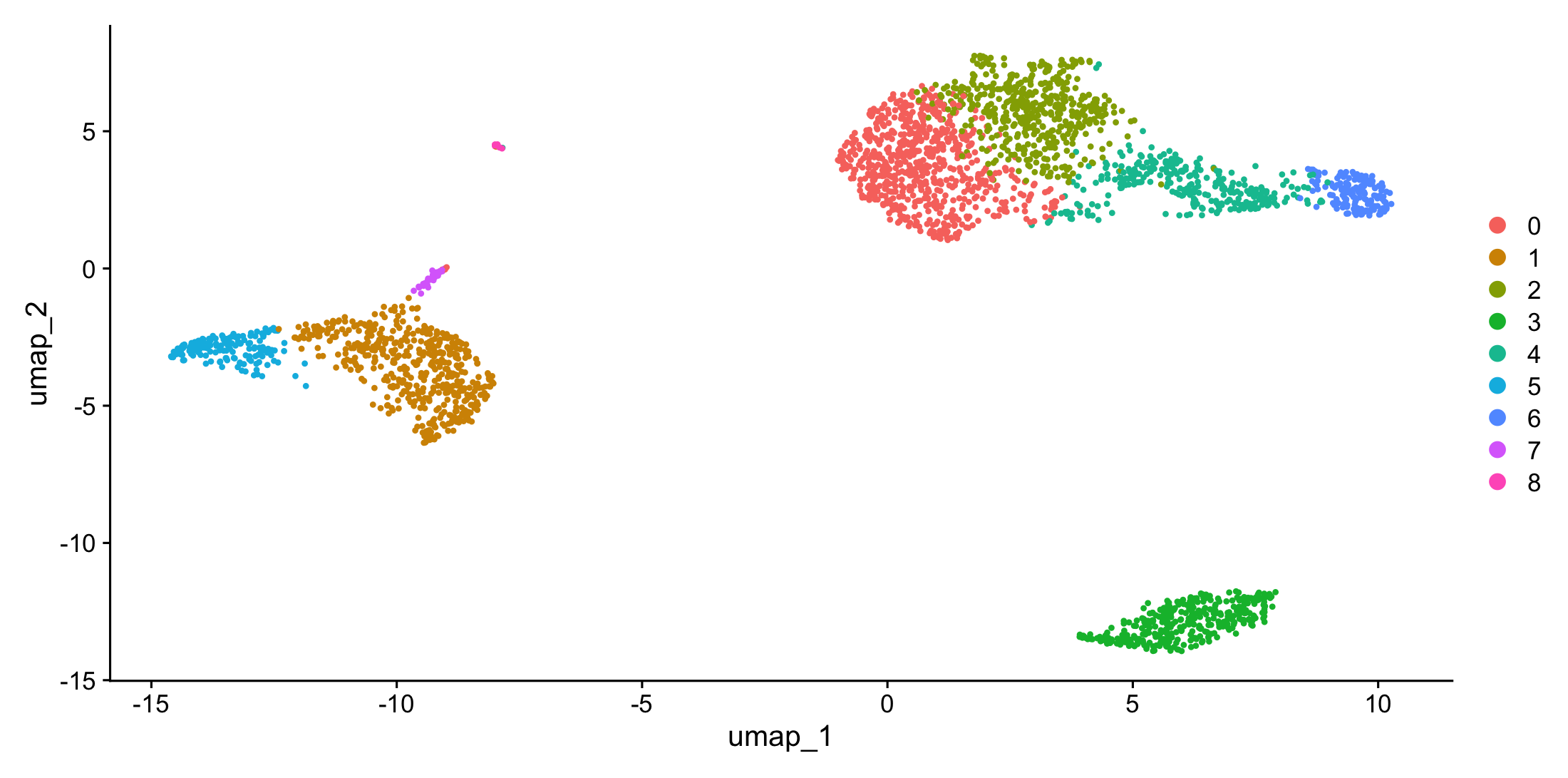
8. Finding Biomarker
각 클러스터가 생물학적으로 어떤 Cell Type 인지 알아내기 위해, 해당 클러스터에만 특이적으로 많이 발현한 유전자를 찾는다.
# 모든 클러스터를 돌며 바이오마커를 발견한다.
pbmc.markers <- FindAllMarkers(pbmc,only.pos = TRUE)
# 발현량이 증가한 유전자만 찾는다.
pbmc.markers %>% group_by(cluster) %>% dplyr::filter(avg_log2FC > 1)
# 특정 마커의 Violin Plot
VlnPlot(pbmc, features = c("MS4A1", "CD79A"))
# you can plot raw counts as well
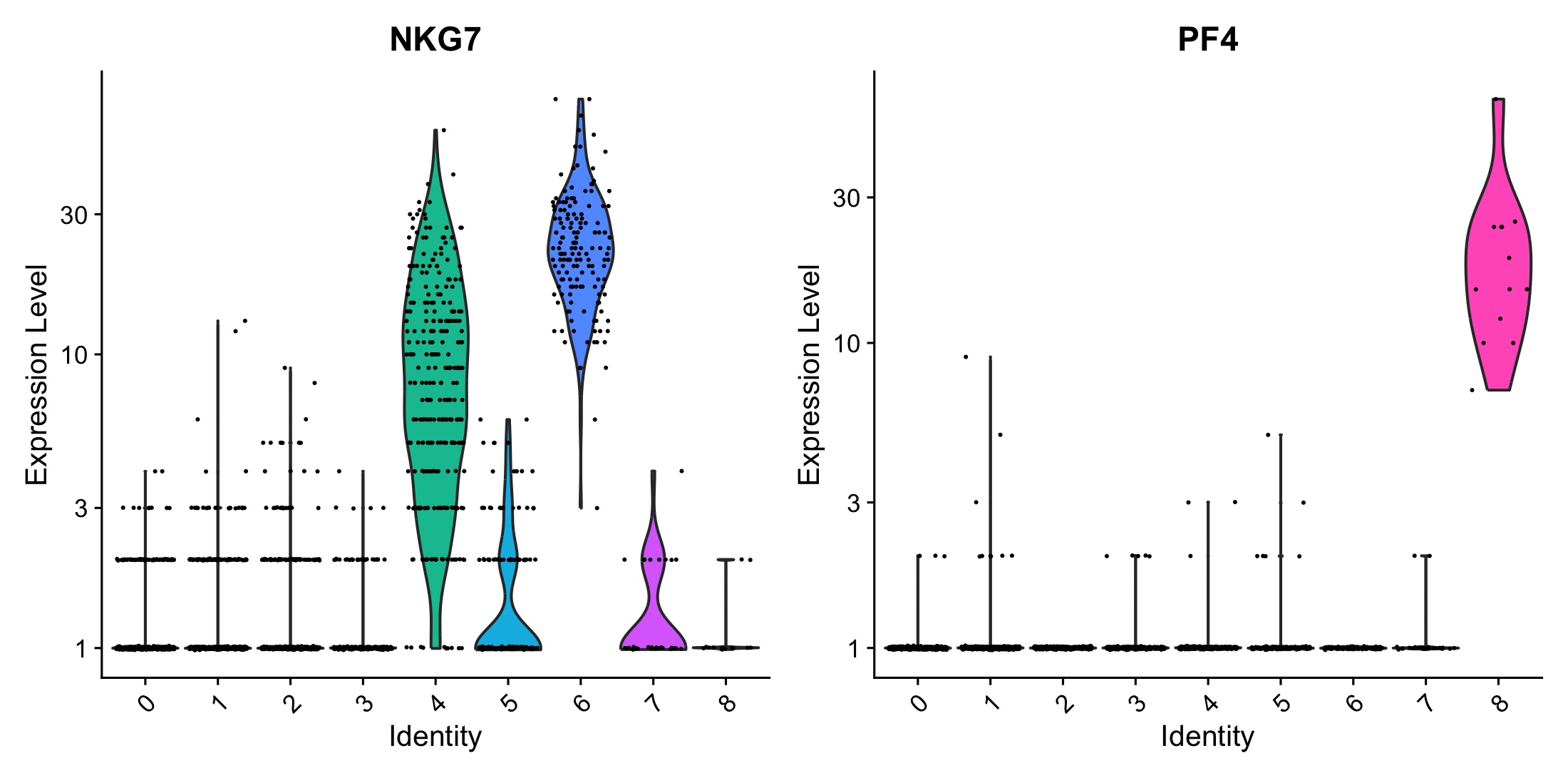
VlnPlot(pbmc, features = c("NKG7", "PF4"), slot = "counts", log = TRUE)
FeaturePlot(pbmc, features = c("MS4A1", "GNLY", "CD3E", "CD14", "FCER1A", "FCGR3A", "LYZ", "PPBP","CD8A"))
# Heatmap
pbmc.markers %>%
group_by(cluster) %>%
dplyr::filter(avg_log2FC > 1) %>%
slice_head(n = 10) %>%
ungroup() -> top10
DoHeatmap(pbmc, features = top10$gene) + NoLegend()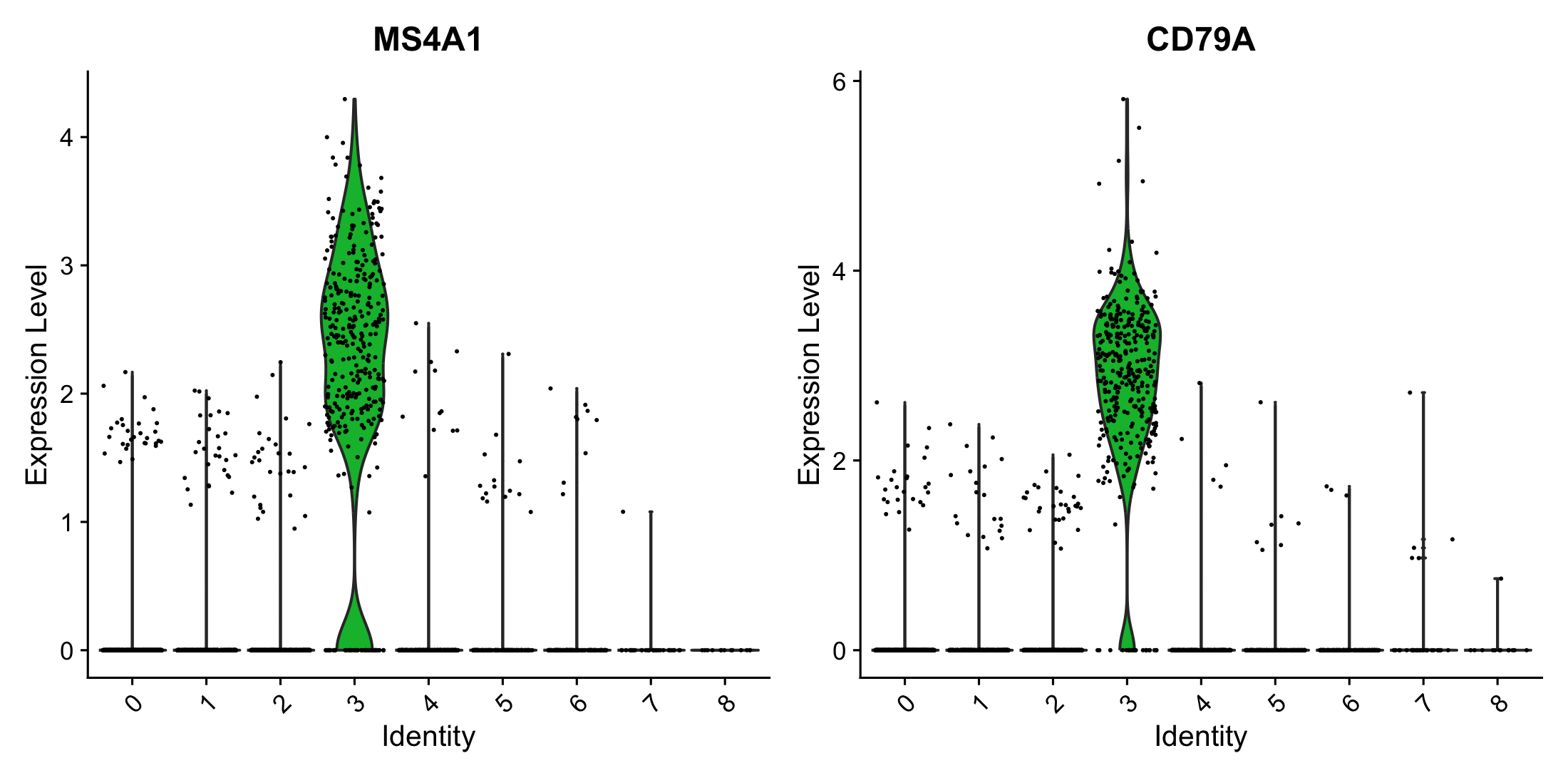
MS4A1 & CD79A는 면역 세포 중 B세포 에서만 특이적으로 발현되는 마커이다.cluster 3에서 특징적으로 분포함을 알 수 있어,cluster 3는 B세포임을 알 수 있다.
-
NKG7은 세포를 죽이는 독성 물질 유전자로, NK세포 혹은 CD8+ T세포에서 주로 나온다.- 주로 4,5,6번 클러스터에서 높게 나타나는데, 그중
cluster 6이 아주 높고 밀도가 높아 NK 세포임을 알 수 있으며,cluster 4가 CD8 T세포일 가능성이 높으나 정확한 구분을 위해 추가적인 마커 확인이 필요하다.
- 주로 4,5,6번 클러스터에서 높게 나타나는데, 그중
-
PF4는 혈소판 인자로,cluster 8에서 높은 분포를 가지므로, 혈소판임을 알 수 있다.
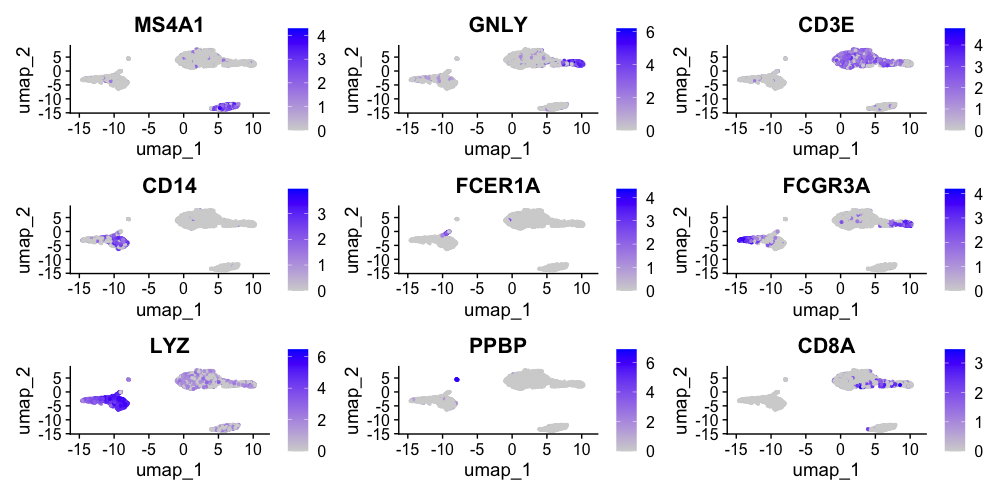
이는 마커들이 위에서 봣던 UMAP상에서 어디에 위치하는가를 표현하는 Feature Plot이다.
CD3E는 T Cell의 마커인데, plot을 보았을때, 우측 상단의 영역에 많이 분포함을 알 수 있다.
이런식으로 생물학적 특징이 기하학적 위치와 어떻게 매칭되는지 알 수 있다.
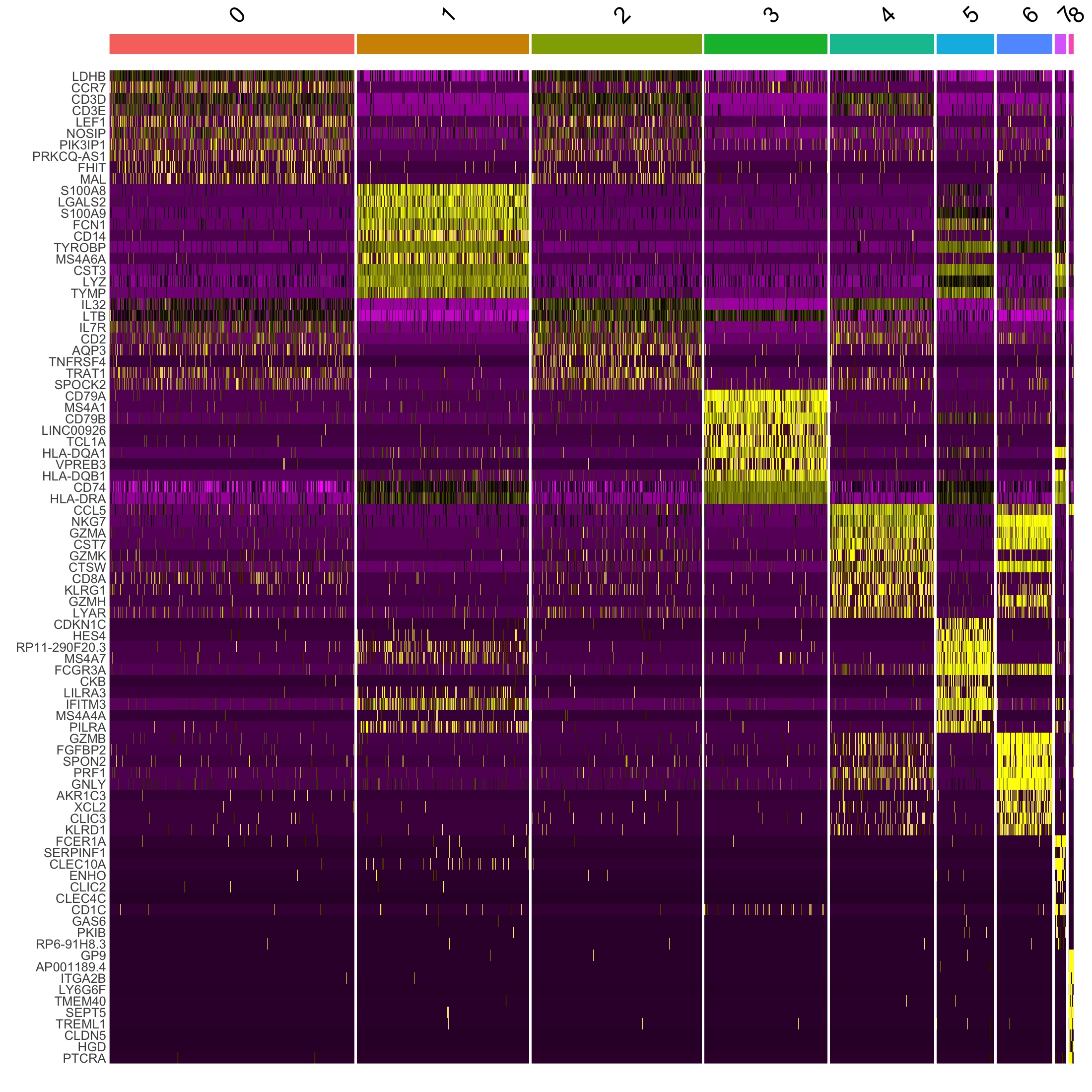
이는 Heatmap으로 모든 분석의 최종 기록이라고 볼 수 있다.
- 가로축 : Cell이며, 위쪽의 숫자가 클러스터 번호이다.
- 세로축 : Gene이며, 각 클러스터를 대표하는 Top10 마커 유전자들이다.
- 노란색 : 유전자 발현량이 높음
- 보라색/검은색 : 유전자 발현량이 낮음 혹은 없음
- 읽는법
cluster0,2의 경우 최상단의LDHB,CD3D,CD3E등 T세포의 마커들의 유전자 발현량이 높다. 전반적으로 비슷한 경향성을 가지고 있지만,CCR7마커의 강도차이 등으로 인해 알고리즘이 다른 클러스터로 분리해둠을 알 수 있다. (cluster0이 Naive T,cluster 2가 Memory T로 분리된다.)cluster 1의 경우 전형적인 단핵구(Monocyte)의 패턴이다.Cluster 3의 경우 확실한 B세포이다.
- 위 히트맵과 같이 노란 영역이 대각선에 집중된다면 각 클러스터가 서로 겹치지 않는 고유한 특징을 가지고 있다는 뜻이다.
9. Annotation
마지막으로, 찾아낸 마커 유전자를 마탕으로 클러스터에 이름표를 붙여준다.
# cell type identity to cluster
new.cluster.ids <- c("Naive CD4 T", "CD14+ Mono", "Memory CD4 T", "B", "CD8 T", "FCGR3A+ Mono",
"NK", "DC", "Platelet")
names(new.cluster.ids) <- levels(pbmc)
# 숫자로 된 cluster ID를 실제 세포 유형 이름으로 변환
pbmc <- RenameIdents(pbmc, new.cluster.ids)
DimPlot(pbmc, reduction = "umap", label = TRUE, pt.size = 0.5) + NoLegend()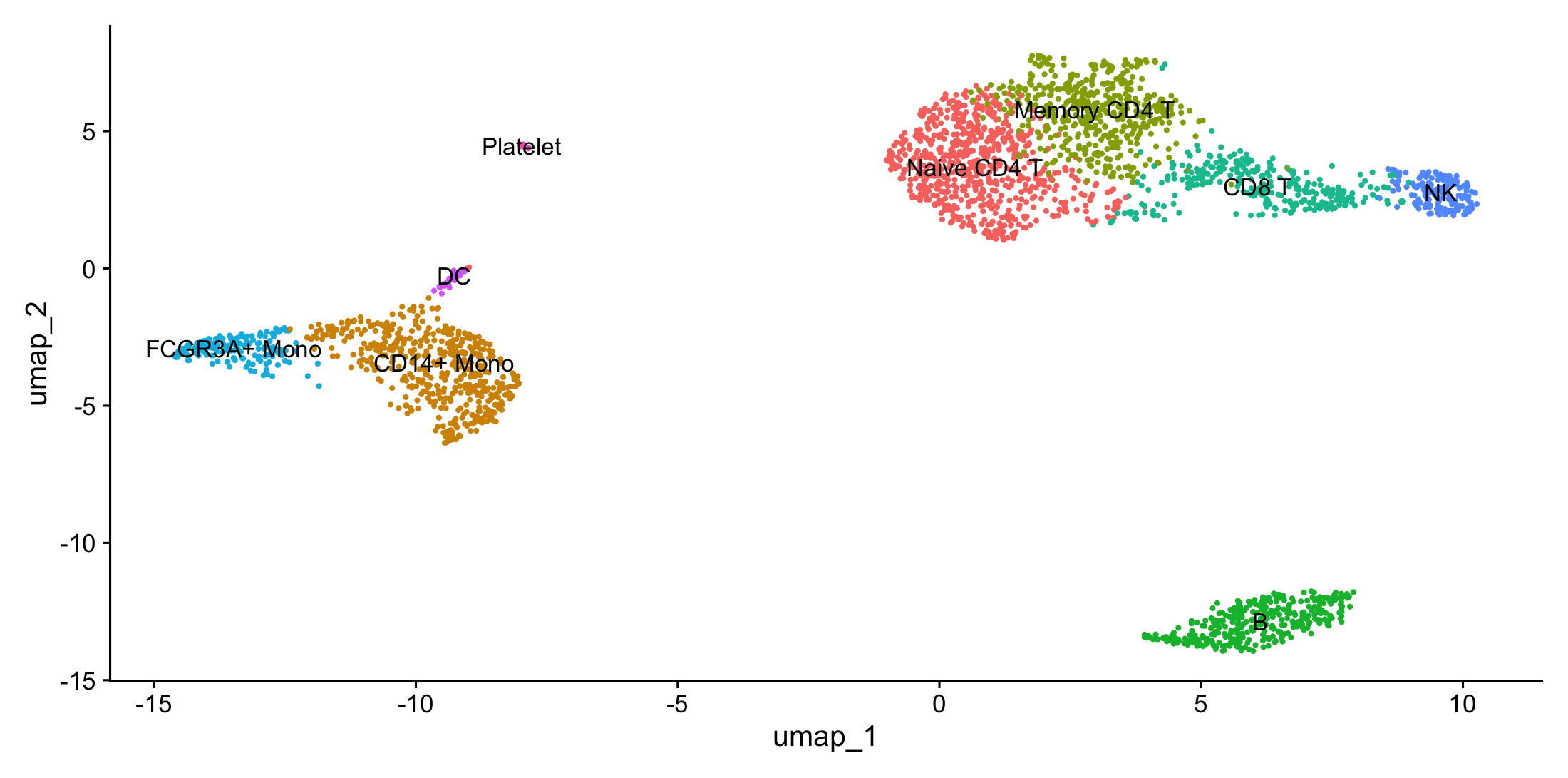
RenameIdents함수를 이용하여 숫자 ID를 실제 이름으로 바꿔줌을 알 수 있다.
