1. FASTQ format
차세대 염기 서열 분석 (Next-Generation Sequencing, NGS) 기술의 발전으로 방대한 양의 유전체 데이터를 얻게 되었는데, 이 데이터를 저장하는 방식이 FASTQ이다.
@SEQ_ID (1. 시퀀스 고유 ID)
GATTTGGGGTTCAAAGCAG... (2. 염기 서열 (A, T, C, G))
+ (3. 구분자)
!''*((((***+))%%%++... (4. 염기 서열의 품질 점수 (Phred Score))Phred 품질 점수
FASTQ 파일의 네 번째 줄의 기호들은 염기 서열의 각 자리가 얼마나 정확한지에 대한 신뢰도 점수이다. 이 점수는 해당 염기가 잘못 분석되었을 확률(P, error probability)를 바탕으로 계산된다.
| Phred 점수(Q) | 에러 확률 | 정확도 |
|---|---|---|
| 10 | 10% | 90% |
| 20 | 1% | 99% |
| 30 | 0.1% | 99.9% |
| 40 | 0.01% | 99.99% |
Phred 점수가 높을수록 해당 염기의 신뢰도가 높다는 의미이다. 일반적으로 Phred 점수 30 이상을 고품질 데이터의 기준으로 삼는다.
2. QC 과정
Quality Control의 핵심 원리는 기준에서 벗어나는 데이터를 찾아내고 원인을 파악하는 것이다.
1. 품질 지표 확인 (Quality Metrics)
FastQC와 같은 툴을 사용해서 데이터의 다양한 품질 지표를 시각적으로 확인한다.
- 전반적인 염기 서열 품질은 괜찮은가? (Per base sequence quality)
- GC 함량 분포는 정상적인가? (Per sequence GC content)
- 중복된 Read가 비정상적으로 많지는 않은가? (Sequence Duplication Levels)
- 시퀀싱 과정에서 사용된 어댑터(Adapter) 서열이 남아있지는 않은가? (Adapter Content)
2. 문제점 확인
- 품질이 낮은 염기 서열이 있다면 -> Trimming(잘라내기)
- 어댑터 서열 발견 -> Adapter Trimming(어댑터 제거)
- 오염 의심 -> 원인 파악 및 오염 서열 제거
3. 데이터 정제 (Data Cleaning)
Trimmomatic,fastq와 같은 툴을 사용하여 실제 문제점을 해결한다.
4. 정제 후 QC (Post-Cleaning QC)
정제된 데이터를 다시 FastQC를 실행하여 문제가 해결되었는지 확인한다
해당 QC과정을 거쳐 신뢰할 수 있는 데이터를 가지고 다음 단계인 Alignment(정렬) 분석을 실행한다.
3. macOS 터미널에서 FastQC 실습하기
# FastQC 설치
brew install fastqc
# 데이터 다운로드
prefetch SRR1553606
fastq-dump --split-files SRR1553606
# fastQC 실행
fastqc SRR1553606_1.fastq SRR1553606은 효모(Saccharomyces cerevisiae)의 RNA - Seq 데이터이다.
해당 데이터를 가지고 fastQC를 실행하면 html파일이 생기는데, 해당 파일을 열어 분석 결과를 볼 수 있다.
4. FastQC Report
우선 각 항목의 지표들이 무슨 의미를 가지는지 알아보자.
Basic Statistics
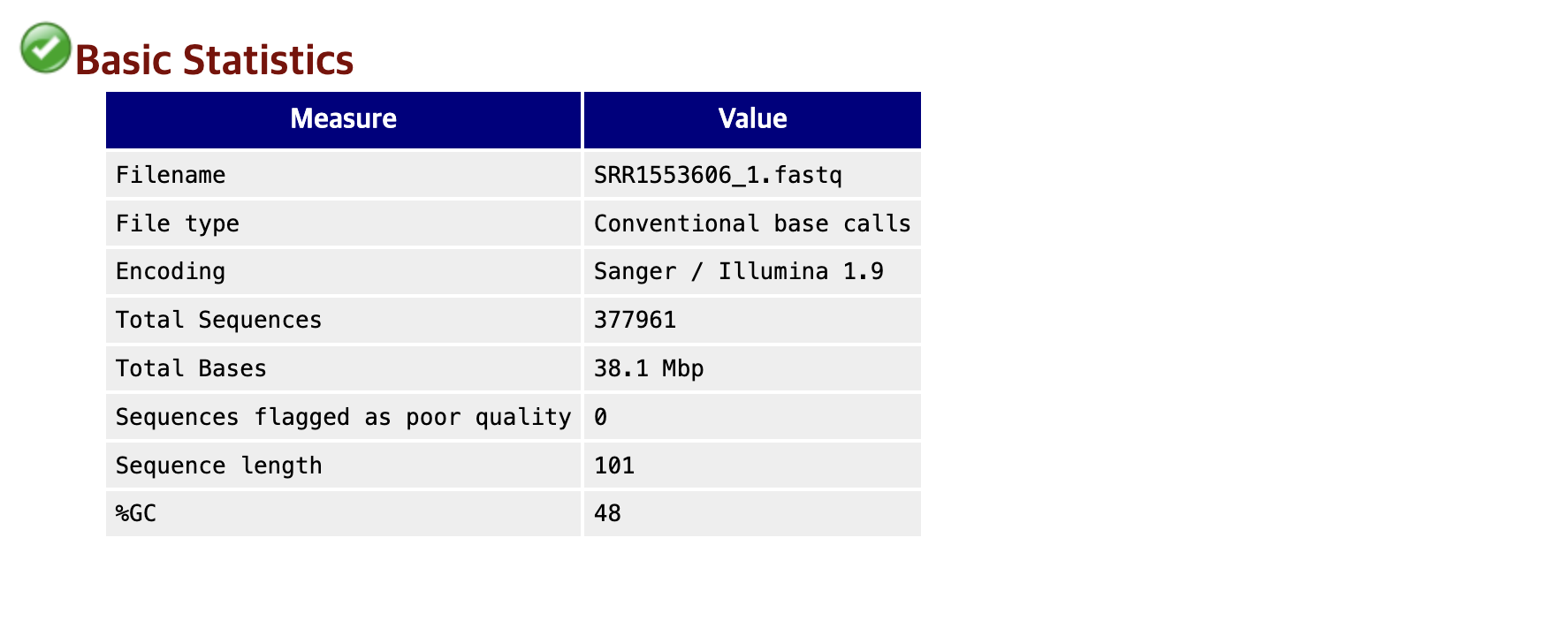
- Total Sequences :Read의 총 개수(FASTQ 파일의 4줄짜리 한 세트가 Read 1개 이다.)
- Total Bases : 총 염기의 수 (약 3,810만개의 염기가 존재)
- Sequences flagged as poor quality : 품질 저하 서열 수 (0은 품질이 심각하게 나쁜 Read는 없다는 의미로, 좋은 결과이다.)
- Sequence length : Read 하나의 염기 서열 길이 (101로 표시된 것은 모든 Read의 길이가 101bp로 동일하다는 의미이다.)
- %GC : 전체 염기중 구아닌(G)와 사이토신(C)이 차지하는 평균 비율. 이 값은 생물 종의 유전체 특성을 반영한다.
Per base sequence quality ❌
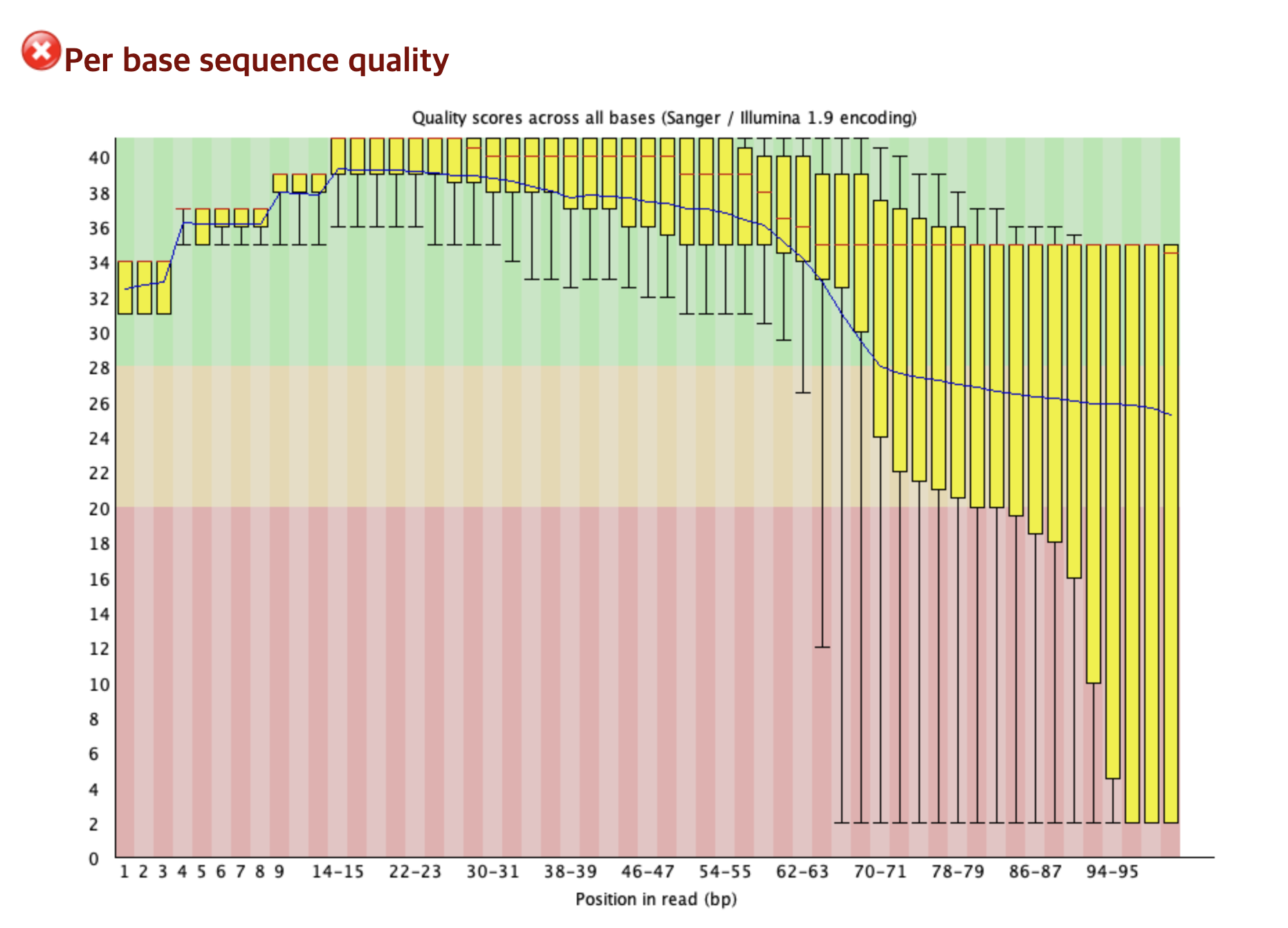
해당 지표는 QC에서 가장 중요한 부분 중 하나이며, 각 위치에서의 Phred 품질 점수의 분포를 박스 플롯으로 설명하고 있다.
결과 분석
- 초반부 ✅ (1 ~ 15bp) : 품질이 약간 흔들리지만, 전반적으로 좋음. 이는 시퀀싱 초반에 나타날 수 있는 자연스러운 현상이다.
- 중반부 ✅ ( 15 ~ 65bp) : 품질이 매우 높고 안정적이다. 대부분의 Read들이 Phread 36 이상인 최상위 품질을 가진다.
- 후반부 ❌ ( 65bp ~ ) : 품질이 급격하게 저하하여 대부분의 염기들을 신뢰할 수 없다.
데이터의 후반부의 품질 저하가 심하므로 Trimming 작업이 필요함을 짐작할 수 있다.
Per sequence quality scores ✅
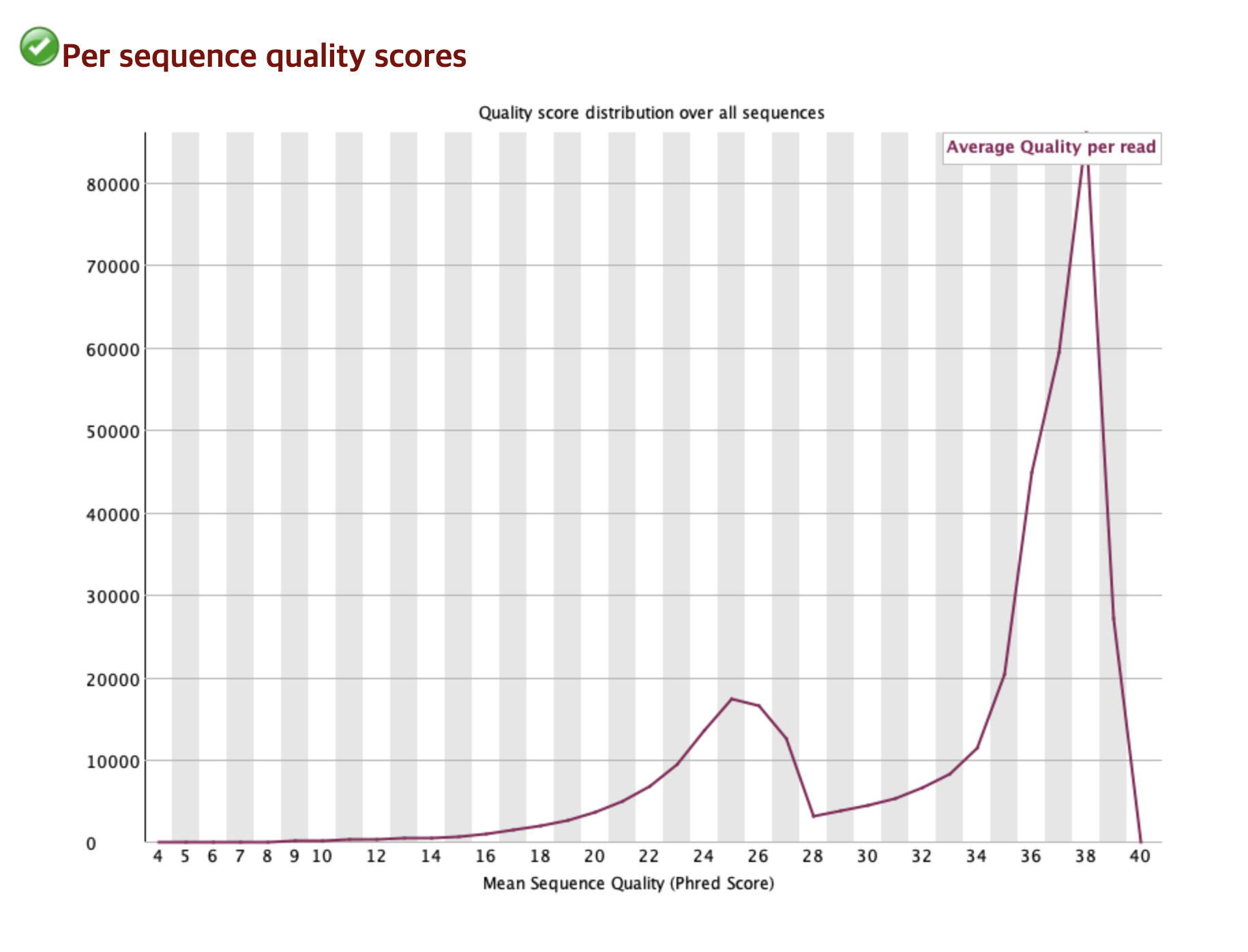
해당 지표는 개별 Read의 평균 품질 점수 분포를 보여준다. 데이터의 전반적으로 품질이 좋음을 알 수 있다.
Per base sequence content ❌
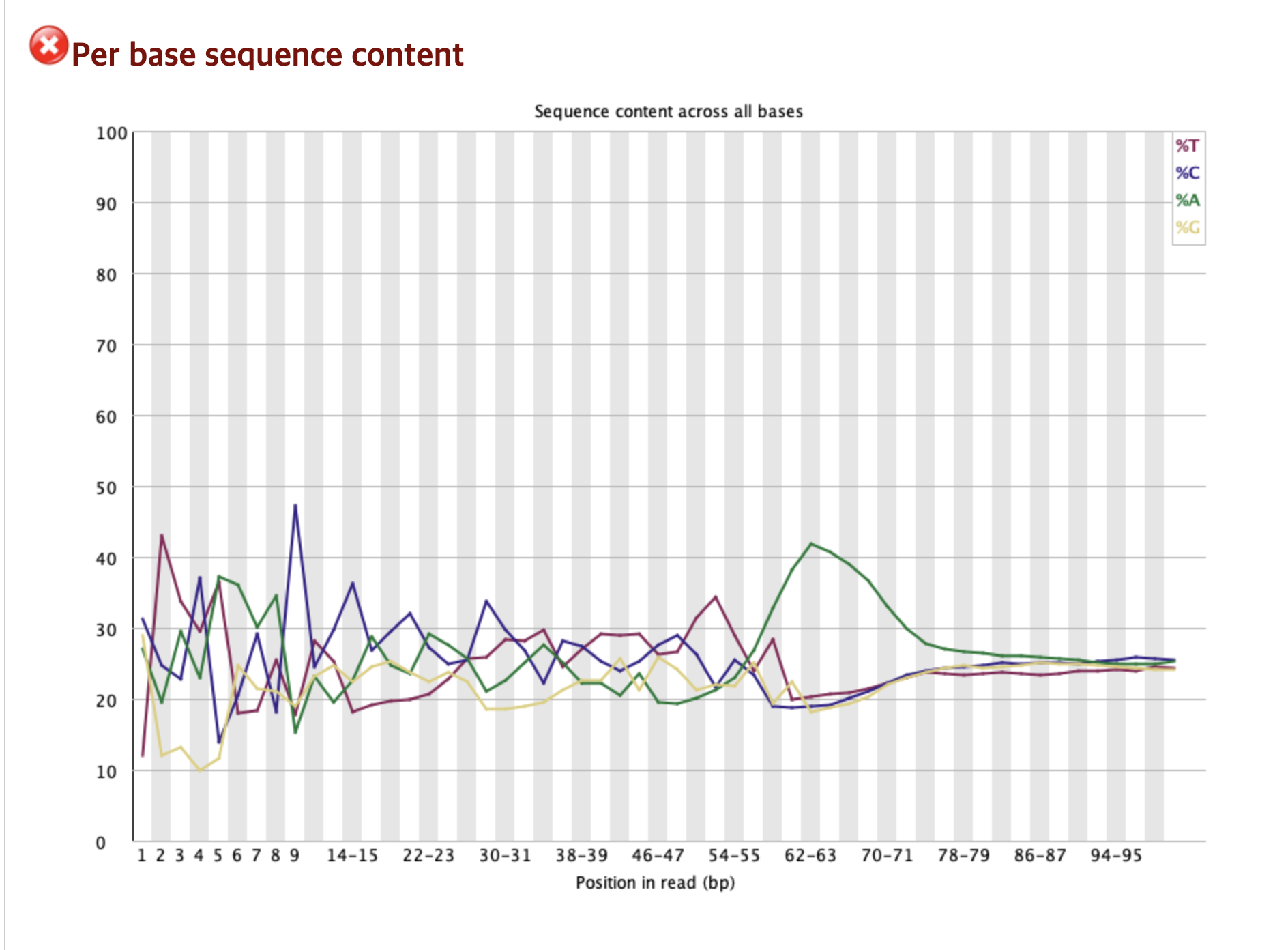
이 그래프는 Read의 각 자리마다 네 종류의 염기가 얼마나 차지하는지 보여준다.
이상적으로는, 4개의 염기가 모든 위치에서 거의 동일한 비율을 차지하여 4개의 선이 평행하게 그려지는 것이다.
결과 분석
초반부의 심한 편향(1 ~ 15bp) 에 의해 해당 그래프의 초반부의 요동이 심한 것을 볼 수 있다. 이는 시퀀싱 초반 염기 서열이 특정 염기들로 쏠려있다는 염기 구성 편향(sequence bias) 가 있다는 강력한 증거이다.
이러한 현상은 RNA-Seq 실험에서 매우 흔하게 나타나는데, 대표적인 원인은 라이브러리 제작 과정에서 사용하는 Random Hexamers(무작위 6개 염기 조합 프라이머) 때문이다.
Per sequence GC content ⚠️
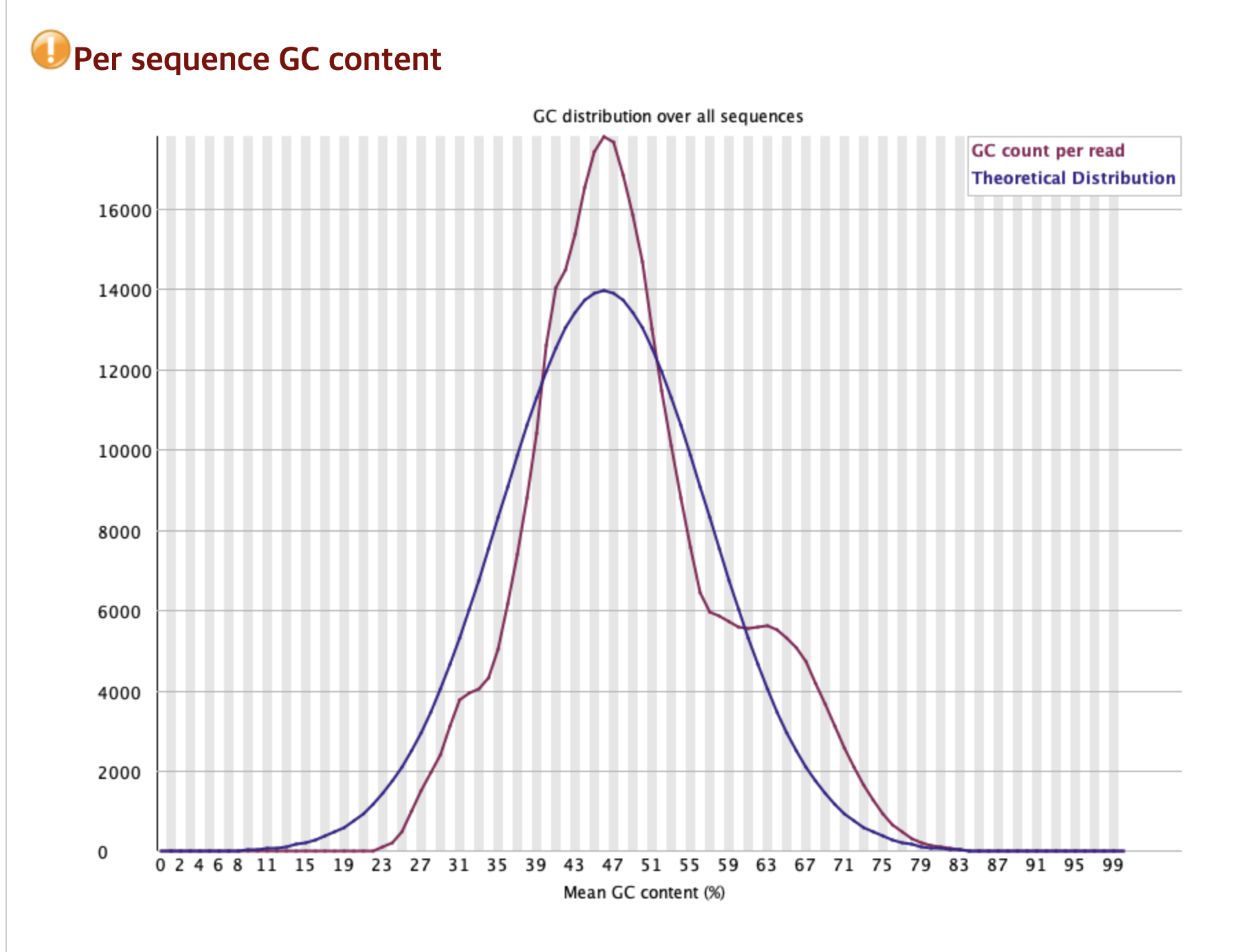
이 그래프는 Read들의 평균 GC함량 분포를 나타낸다. 빨간 선이 실제 데이터 분포이고, 파란 선은 컴퓨터가 계산한 이상적인 정규 분포 모양이다.
결과 분석
파란 색 선이 빨간색 선과 완전히 일치하지 않고, 모양이 약간 비정상적이기 때문에 ⚠️ 표시가 되었다.
주 봉우리 오른쪽(60-65%)근처에 작은 봉우리가 하나 보이는 것이 문제임을 알 수 있다.
이러한 비정상적인 분포는 오염이거나 생물학적 특성임을 시사한다.
Per base N content ✅
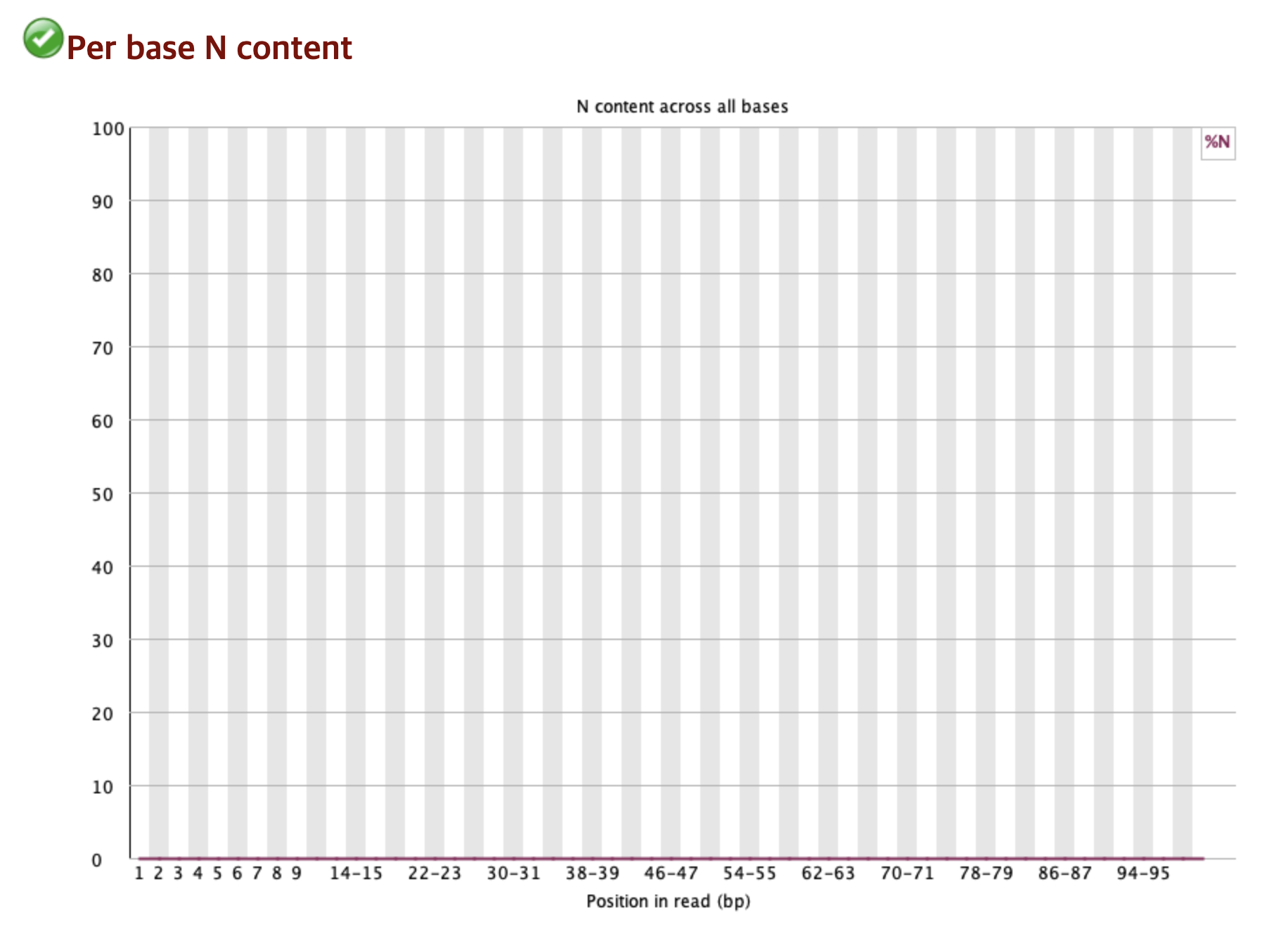
시퀀싱 장비가 특정 위치의 염기를 특정할 수 없을때 그 자리를 N으로 표시하는데, 그러한 N의 비율을 나타낸 그래프이다.
해당 데이터는 N이 완벽하게 없으므로, 어떠한 위치에서도 염기를 판독하지 못한 경우가 없었다라는 의미이다.
Sequence Length Distribution ✅
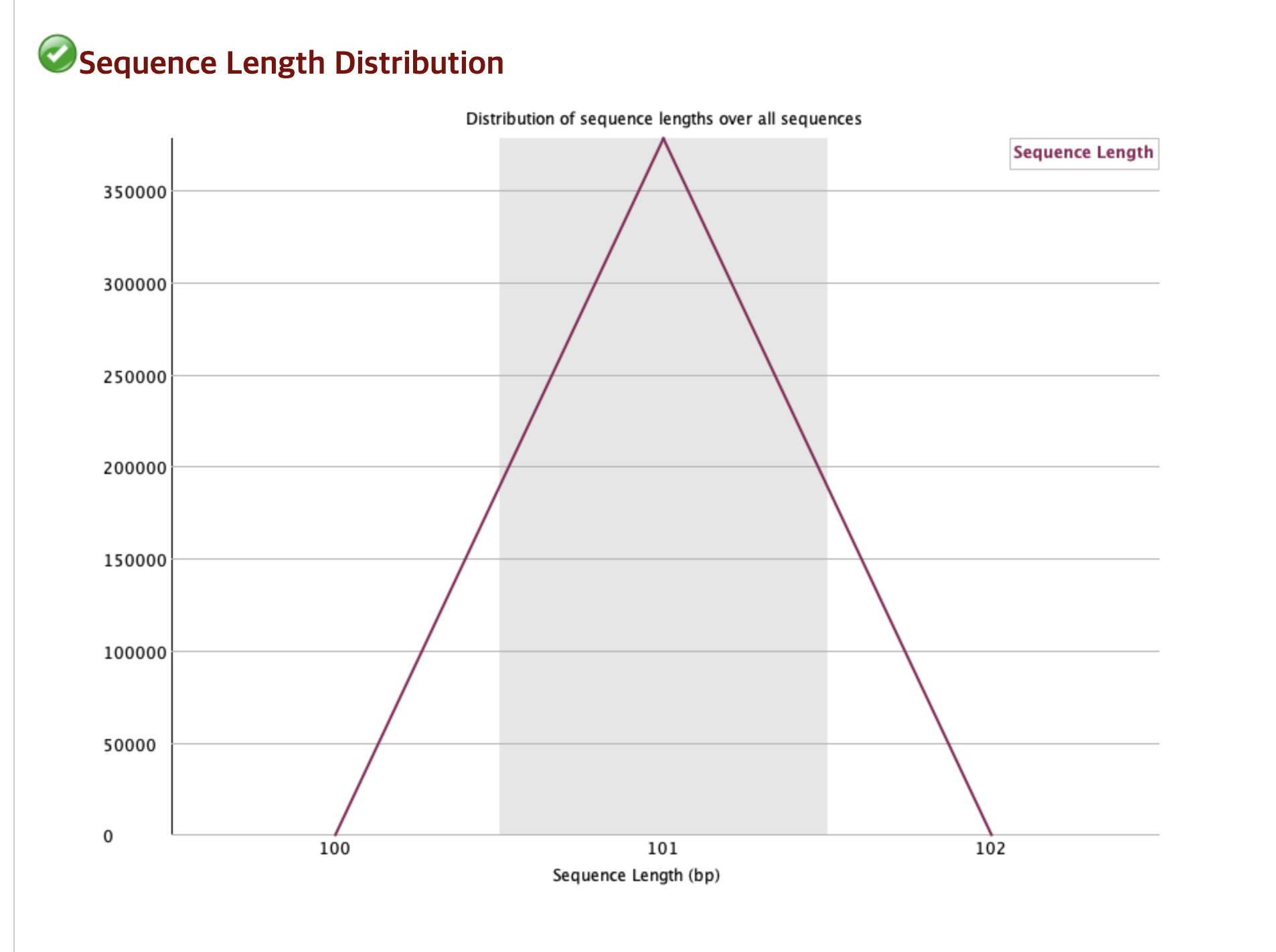
서열 길이 분포를 나타내며, 해당 데이터의 모든 Read의 길이가 101bp로 동일하다는 좋은 결과를 나타낸다.
대부분의 illumina 시퀀싱 장비는 길이가 고정된 데이터를 생성하기 때문에, 하나의 길이만이 나타나는 그래프가 가장 이상적이다.
Sequence Duplication Levels ❌
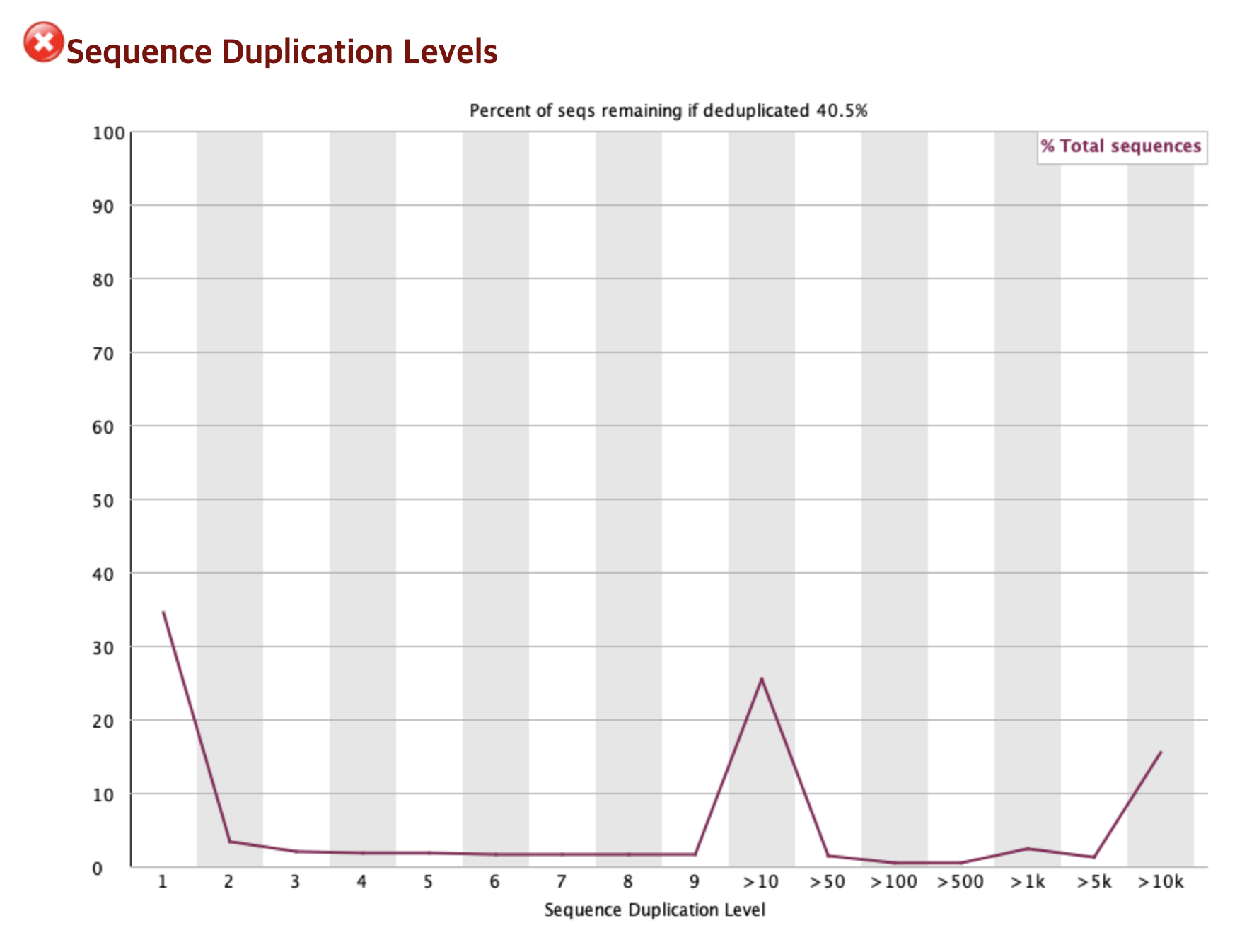
이 항목은 ❌ 가 표시되었지만 RNA-Seq 데이터에서는 지극히 정상적인 결과일 수 있기 때문에 해석에 주의가 필요하다.
이 그래프는 완전한 동일한 서열이 데이터에 얼마나 많이 중복되었는지 나타낸다.
RNA-Seq는 유전자 발현량을 측정하는 실험인데, 세포 내에는 발현량이 매우 높은 유전자(Housekeeping gene)와 발현량이 낮은 유전자가 공존한다. 따라서 발현량이 매우 높은 유전자에서 유래한 RNA 서열은 당연하게도 데이터에 수만 번 이상 중복되어 나타난다.
Overrepresented sequences ❌
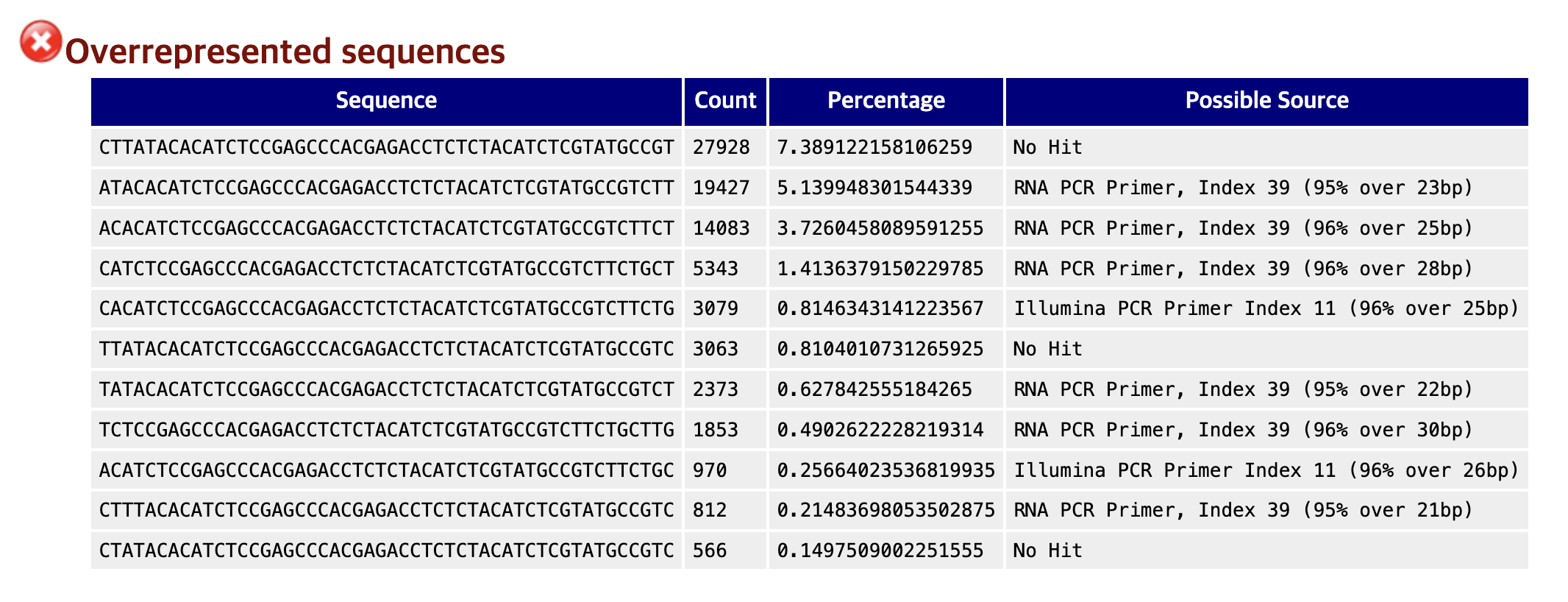
이 표는 데이터에 비정상적으로 많이 나타내는 서열들의 목록이다.
'Possible Source' 항목을 보면 FastQC가 이 서열들의 정체를 RNA PCR Primer또는 Illumina PCR Primer 등으로 정확히 찾아낸 것을 볼 수 있다. 이들은 모두 라이브러리 제작에 사용되는 일종의 어댑터 서열이다.
Adapter Content ❌
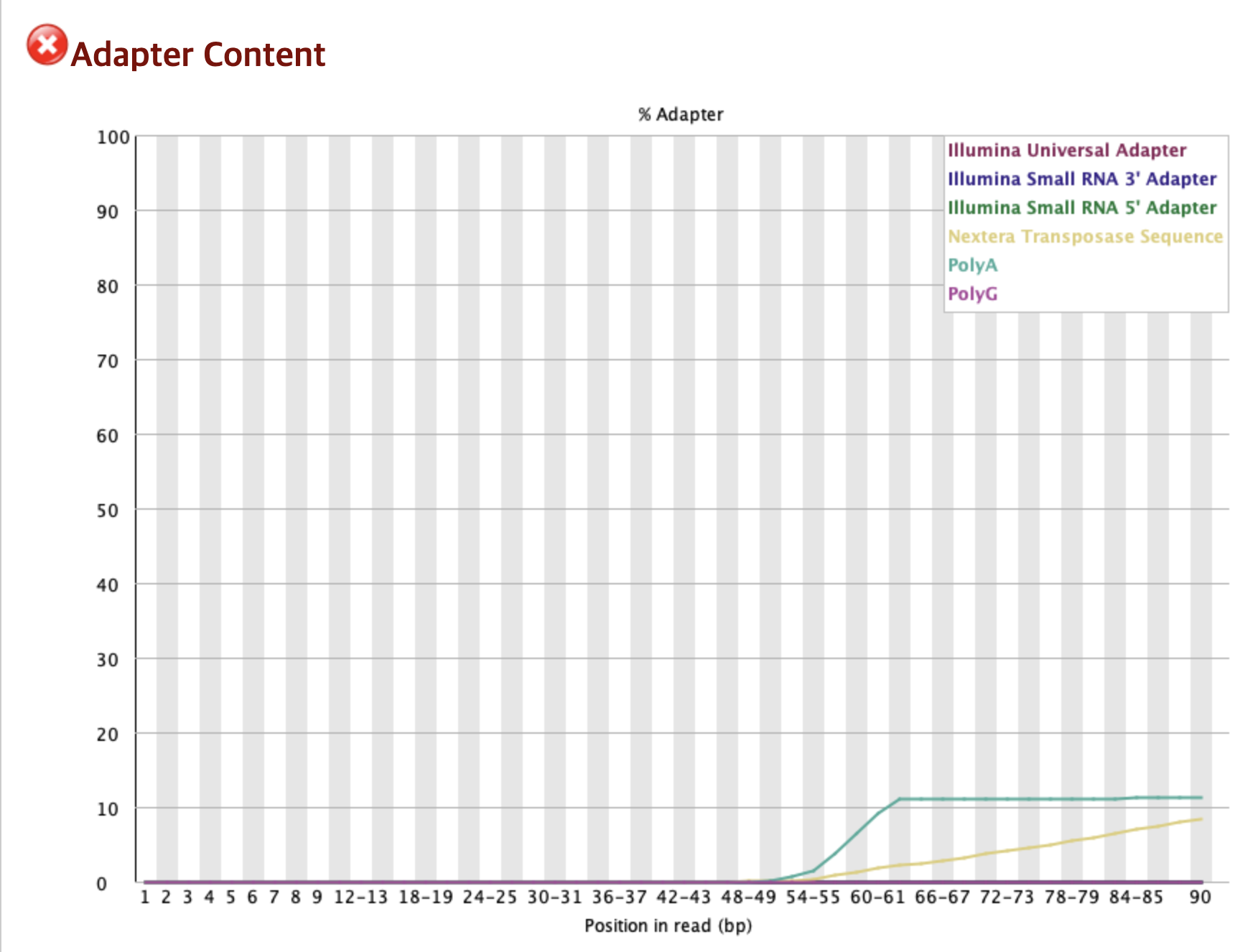
이 그래프는 Read의 위치에 따라 Adapter 서열이 얼마나 포함되었는지 나타내는데,
그래프를 보면 Read의 뒷부분(약 55bp 이후)으로 갈수록 어댑터 서열의 비율이 급격하게 증가하는 것을 볼 수 있다.
이는 실제 분석 대상인 RNA 조각의 길이가 Read길이(101bp)보다 짧아 시퀀싱 장비가 RNA 조각을 다 읽고 그 이후의 Adapter 서열을 읽었음을 알 수 있다.
그러므도 해당 서열을 Trimming을 통해 반드시 제거 해야함을 알 수 있다.
정리를 해보자면 ..
첫번째 QC 결과를 따르면, 우리 데이터에는 여러 문제들이 있었다.
1. Adatper Sequences
2. 후반부의 Low Quality bases
다음 문제들을 위해 Trimming을 해보도록 하자.
5. Trimming
# Trimming 을 위한 라이브러리 설치
brew install fastq
fastp \
-i SRR1553606_1.fastq \
-I SRR1553606_2.fastq \
-o trimmed.SRR1553606_1.fastq \
-O trimmed.SRR1553606_2.fastq \
-h trimmed.report.html
# Trimming 후 post - QC 실행
fastqc trimmed.SRR1553606_1.fastq-i,-I: 입력 파일-o,-O: 출력 파일-h: Trimming 결과에 대한 리포트 생성
다음 과정을 걸치고 나면 trimmed. 접두사가 붙은 fastq 파일과 trimmed.report.html파일이 생김을 알 수 있다.
6. 결과
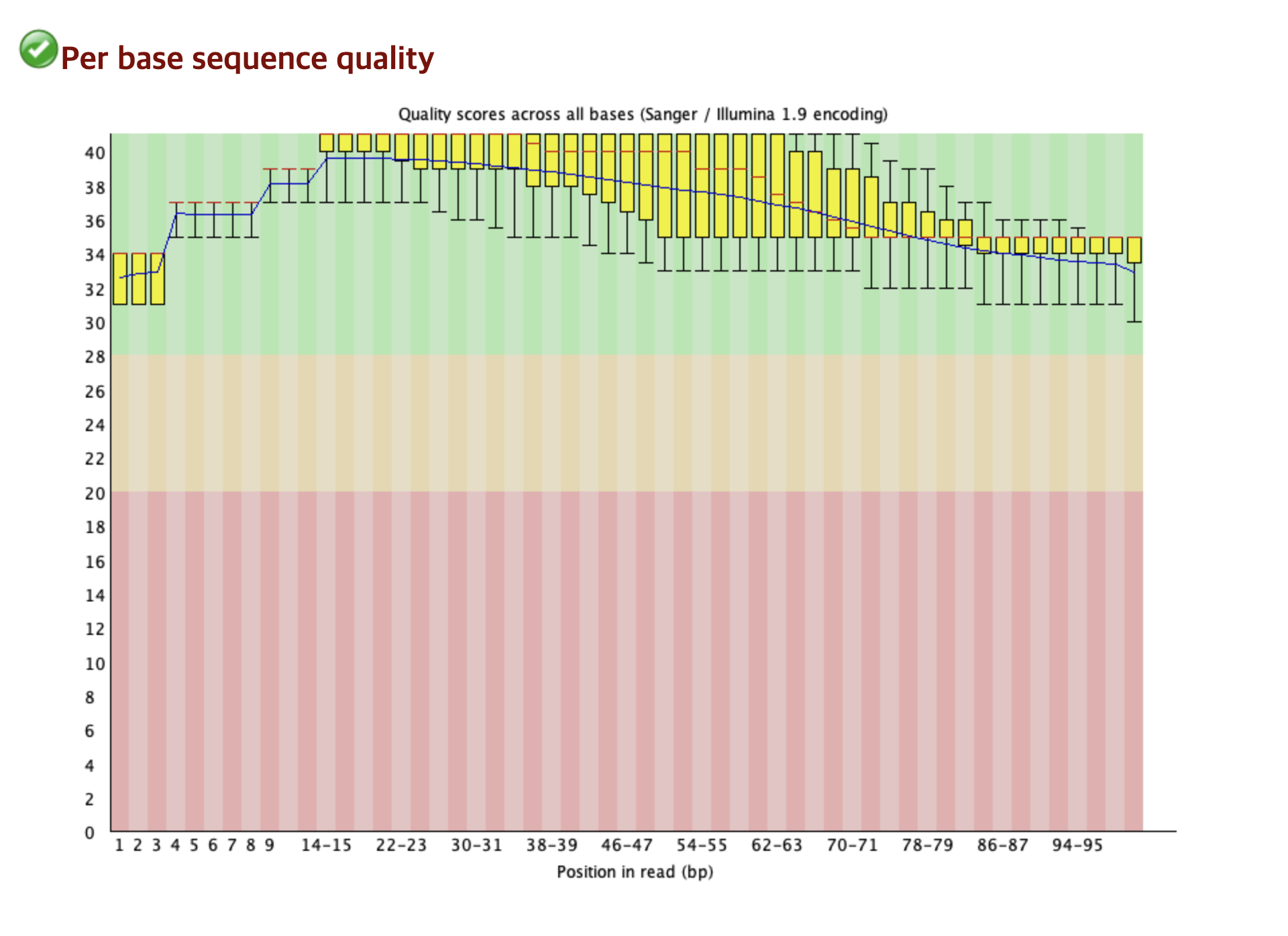
아까의 Per base sequence quality 그래프와 비교해보면 후반부의 저품질의 데이터가 삭제되어 좋은 품질의 데이터만이 남음을 알 수 있다.
즉, 분석에 방해가 되던 서열들이 제거 되며 데이터의 품질이 극적으로 향상되었음을 알 수 있다.
상태가 바뀐 항목들
- Per base sequence quality (염기 서열 품질): (❌ Fail → ✅ Pass)
Read 뒷부분의 품질이 좋지 않은 염기들이 성공적으로 잘려나가 전체적인 품질이 향상 되었다. - Adapter Content (어댑터 포함): (❌ Fail → ✅ Pass)
분석을 방해하던 어댑터 서열이 제거되었다. - Overrepresented sequences (과대 발현 서열): (❌ Fail → ✅ Pass)
어댑터 서열이 사라지면서 비정상적으로 많던 서열 문제가 해결되었다. - Sequence Length Distribution (서열 길이 분포): (✅ Pass → ⚠️ Warn)
Trimming 전에는 모든 Read의 길이가 101bp로 일정했지만, 각 Read의 품질에 따라 서로 다른 길이로 잘려 나갔기 때문에 분포가 다양해 졌다. 이는 Trimming의 정상적인 결과이다. - Per sequence GC content (GC 함량 분포): (⚠️ Warn → ❌ Fail)
Trimming의 결과로 전체 Read들의 GC분포가 달라졌다. 데이터가 나빠진 것이 아니라 구성이 바뀐것이므로 신경쓰지 않아도 된다. - Sequence Duplication Levels (중복 서열 수준): (❌ Fail → ❌ Fail)
이는 RNA-Seq의 유전자 발현량 차이를 의미하므로 Trimming으로 해결되지 않는 문제이다.
이제 Trimming의 결과로 분석을 방해하던 낮은 품질의 데이터, 어댑터문제는 해결되었다. 이 신뢰성 있는 데이터를 가지고 다음 단계인 Alignment(유전체에 서열을 정렬하는 작업) 을 진행하면 된다.
