이번 Bulk seq Analysis 연습은 2024년도 Cell reports에 출간된 논문 데이터를 이용하여 진행하겠습니다.
Bulk seq analysis에서 Data를 처리하는 프로그램은 굉장히 다양한데, 개인적으로 Linux 서버를 이용하는 유저라면 가장 편리하게 사용할 수 있는 tool이 Kallisto program이라고 생각합니다.
때문에 이번 Bulk seq analysis 연습에서는 Kallisto - tximport - Deseq2 를 사용하여 진행하겠습니다.
(Kallisto를 사용하는 이유에 대해서는 추후에 다루도록 하겠습니다)

#Download data
대개 단일세포분석이든 Bulk seq 분석이든, Sequencing 분석을 진행한 논문에서는 Data availability section을 따로 만들어 데이터를 다운받을 수 있는 Accession number를 남깁니다.
데이터는 National Center for Biotechnology Information(NCBI)의 Gene Expression Obnibus(GEO) 데이터 베이스에서 다운받을 수 있습니다.

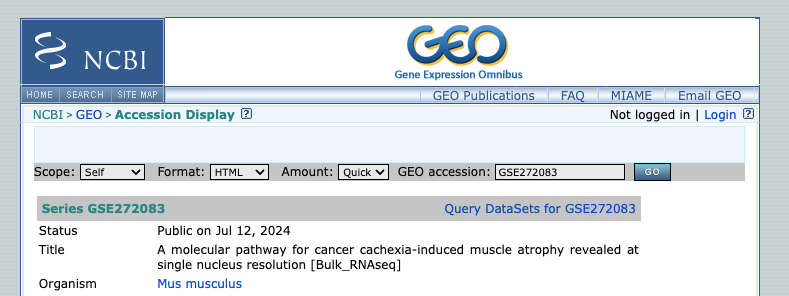
Geo에 들어가서 accession number를 입력하면 다음과 같은 창이 뜹니다.
위에 부분에는 출간된 paper의 제목과 seq data 형태에 대한 정보가 있고,
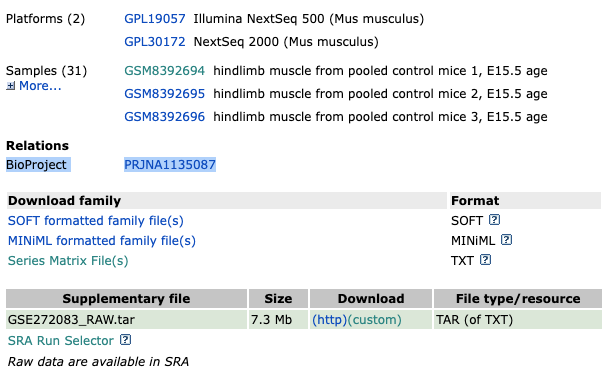
아래 부분에는 실제 데이터 파일들이 있습니다.
여기서 저희가 필요한 것은 Bioproject 란에 적혀있는 project number (PRJNA1135087) 입니다.
SRA Explorer 사이트에 들어가서 복사된 Project number(PRJNA1135087)를 검색해줍니다.
Project number를 검색하면, 하나의 Project에서 사용된 모든 FastQ 파일 형식의 sequencing data들이 나열되어있습니다.
그 목록에서 본인이 원하는 데이터만 클릭하여 Add N to Collection 버튼을 눌러줍니다.
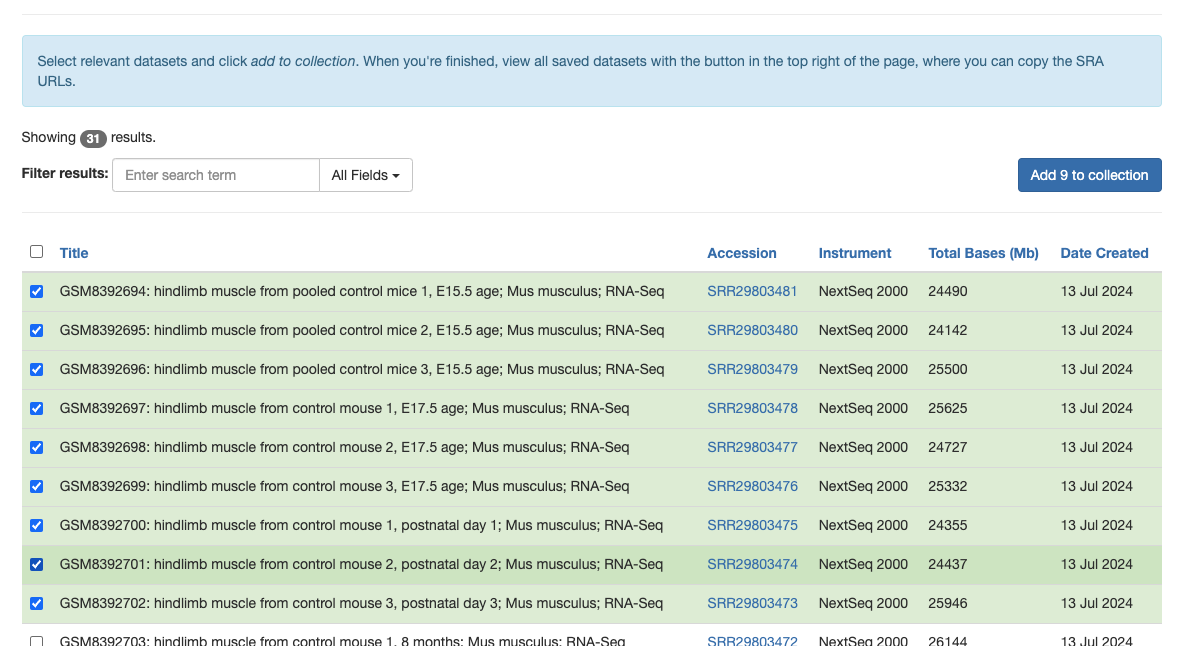
그 후에 오른쪽 상단에 N Saved datasets 버튼을 누르면, 데이터를 다운받을 수 있는 다양한 링크들이 나옵니다.
데이터를 다운받는 방법은 여러 가지가 있지만, 저는 Raw FastQ Download URLs를 이용한 방법을 선호합니다.
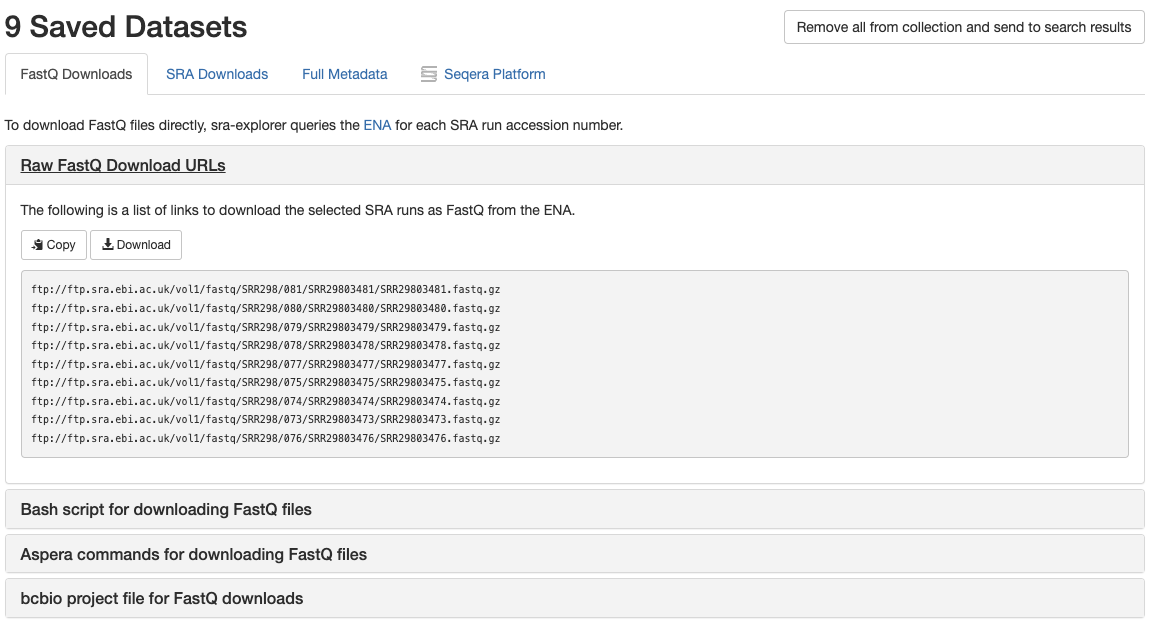
이제 이 링크들을 이용해서 Linux 서버에 데이터들을 다운받습니다.
이때 코드는 다음과 같이 진행하면 됩니다.
(base) Daehwankim:~$ mkdir Bulk_Practice && cd Bulk_Practice
(base) Daehwankim:~/Bulk_Practice$ wget -O Fast1.fq.gz ftp://ftp.sra.ebi.ac.uk/vol1/fastq/SRR298/081/SRR29803481/SRR29803481.fastq.gz여기서 한 가지 TIP을 드리자면, 하나의 데이터 세트 당 하나의 directory를 만들어서 각각의 데이터를 따로 저장하는 것을 추천합니다.
그 이유는 추후에 Kallisto로 Quantification를 진행하여 새로운 파일이 생성될 때,
파일의 이름이 하나로 고정되어 생성되기 때문에, 마치 하나의 파일에 데이터를 덮어쓰기를 하는 것처럼 맨 마지막 파일의 데이터만 남기 때문입니다.
이는 나중에 더 자세히 설명하도록 하겠습니다.
이번 글에서는 Bulk seq Analysis를 위해 Linux 서버에 Public data를 다운받는 방법에 대해서 다루었습니다.
다음 글에서는 데이터 전처리 과정 (Indexing, Mapping, and Quantification)에 대해서 다루도록 하겠습니다.
감사합니다.
