#Pre processing
Step 1. Create New Environment
기존에 Linux 기반의 서버를 사용하는 유저라면 이미 잘 아시겠지만, 하나의 환경(Environment)에서 배타적인 두 개의 프로그램은 구동이 불가합니다.
때문에 Kallisto program을 구동하기 위해 새로운 환경을 만들어줘야 합니다.
코드는 다음과 같이 진행하면 됩니다.
(base) daehwankim:~/Bulk_Practice$ conda activate Kallisto
(Kallisto) daehwankim:~/Bulk_Practice$ conda install bioconda::kallisto이제 새로운 환경에서 Kallisto program을 구동할 준비가 되었으니, 본격적으로 전처리 작업을 진행하겠습니다.
Step 2. Indexing
인덱싱(Indexing)은 참조 유전자 서열(Reference genome or transcriptome)을 효율적으로 검색할 수 있도록 데이터 정렬 형태를 최적화하는 작업이라고 생각하시면 됩니다.
즉, 참조 서열을 빠르게 검색할 수 있도록 최적화하는 작업이 "인덱싱" 입니다.
코드는 다음과 같이 진행하시면 됩니다.
(참고로, Bulk seq analysis에서 사용할 참조 유전자 서열 데이터는 GENCODE 사이트에서 제공하는 데이터를 이용하면 됩니다.)
#Reference gene download
(Kallisto) daehwankim:~/Bulk_practice$ wget -O Referce.Transcriptome.fa.gz https://ftp.ebi.ac.uk/pub/databases/gencode/Gencode_mouse/release_M36/gencode.vM36.transcripts.fa.gz
#Indexing
(Kallisto) daehwankim:~/Bulk_Practice$ Kallisto index -i Indexed_Reference.fa ./Referce.Transcriptome.fa.gz
Step 3. Mapping & Quantification
Mapping은 Sample의 RNA 데이터를 참조 유전자 서열과 비교하는 과정입니다.
즉, 각 read들이 참조 서열의 어느 부분에 해당하는 지 찾고, 이를 통해 read의 서열이 어떤 유전자 혹은 엑손에 속하는 지 결정합니다.
Quantification은 Mapping 결과를 기반으로, 각 유전자에 매핑된 read의 수를 세어 발현량(Counts)을 계산하는 단계입니다.
즉, 어떤 유전자가 얼마나 많이 발현되었는가를 정량적으로 나타내는 단계입니다.
Mapping 및 Quantification은 Kallist quant를 사용하면 한 번에 처리 가능합니다.
Sequencing이 Paired-end(양쪽 모두 읽음)로 진행되었느냐, Single-end(한쪽만 읽음)로 진행되었느냐에 따라 코드 진행은 약간 다릅니다.
이 논문에서 사용된 데이터는 Single-end sequencing이기 때문에 코드 진행은 다음과 같습니다.
#Single-end
(Kallisto) daehwankim:~/Bulk_Practic$ kallisto quant -i /Path_to/indexed_reference.fa -o ./ --single -l 200 -s 20 ./SRR29803451.fastq.gz -t 20
참고로, 여기서 Path_to라고 하는 것은 데이터가 있는 파일 경로를 간단하게 나타낸 것입니다.
실제로 여러분이 따라하실 때에는 본인의 파일 경로 모두를 정확하게 써주셔야 합니다
#Example
kallisto quant -i /home/daehwankim/00_Reference/indexed_reference.fa -o ./ --single -l 200 -s 20 ./SRR29803452.fastq.gz -t 20
여기서 주의해야할 점은 하나의 directory에 하나의 데이터 세트가 들어가게 만든 후 Quntification을 진행해야 한다는 것입니다.
이는 이전 글에서 말씀드렸다시피, Kallisto로 Quantification를 진행하여 새로운 파일이 생성될 때,
파일의 이름이 하나로 고정되어 생성되기 때문에, 마치 하나의 파일에 데이터를 덮어쓰기를 하는 것처럼 맨 마지막 파일의 데이터만 남기 때문입니다.
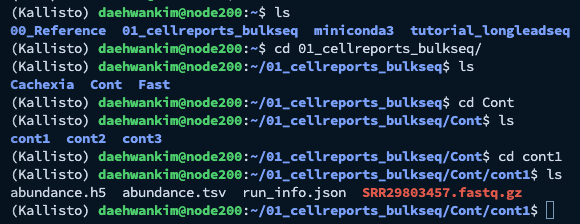
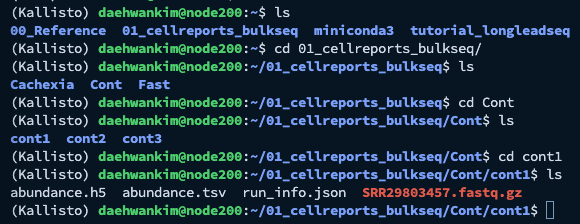
예시로, 저는 'Cont' 라는 directory 안에 cont1, cont2, 그리고 cont3 라는 하위 directory를 만들었습니다.
그리고 각 directory에 하나의 데이터 세트를 넣고 Quantification을 진행하였습니다.
(Kallisto) daehwankim:~/bulk_Practice$ kallisto quant -i ./../Path_to/indexed_reference.fa -o ./Cont/cont1 --single -l 200 -s 20
./Cont/cont1/SRR29803457.fastq.gz -t 20위 코드에서는 평균 read 길이를 200bp로, 표준편차는 20bp로 가정하여 Quntification을 진행합니다.
마지막으로, -t는 Thread 개수를 지정하는 코드인데, 20개의 Thread를 사용해 병렬 처리하겠다는 의미입니다.
때문에 너무 큰 숫자를 쓰면 서버가 감당 못할 수 있으니 20을 넘지 않는 선에서 하시는 것을 추천합니다.
여기까지 잘 따라오셨다면,
다음과 같이 Cont라는 상위 디렉토리 안에 cont1, cont2, 그리고 cont3라는 하위 디렉토리가 만들어져있고,
각각의 하위 디렉토리 안에는 abundance.h5, abundance.tsv 등등의 파일이 들어있는 것을 확인할 수 있습니다.
여기서 저희가 사용할 파일은 abundance.tsv 입니다.
Step 4. Data Export (From server to local)
이제 전처리 과정이 완료된 데이터를 서버에서 로컬(내 컴퓨터의 저장소)로 가져와 R studio에서 사용할 준비를 해야합니다
이때 코드는 scp를 사용하는데, 서버가 아닌 터미널에서 진행해주셔야 합니다.
(base) daehwankim-MacBookAir ~ % scp daehwankim@000.000.000.000:/Path_to/cont1/abundance.tsv /Users/daehwankim/Desktop/Bulk_seq_practice/Cont
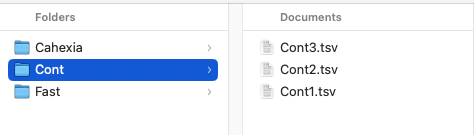
여기까지 잘 따라오셨다면, 총 9개의 파일들이 컴퓨터에 잘 저장된 것을 볼 수 있을 겁니다.
마지막으로 저장된 파일들의 이름을 본인의 입맛에 따라 바꿔주면 다음과 같이 됩니다.
다음 글에서는 Differential Expression Gene (DEG) 분석을 위해 필요한 DESeqDataSet을 생성하는 방법에 대해서 다루도록 하겠습니다.
감사합니다.
