Step1: Reference transcriptome indexing
- kallisto는 transcriptome을 reference로 사용함!
- genecode에서 다운받은 transcriptome을 indexing해주는 작업이 필요하다.
- non coding 부분을 제외하고 싶다면 protein coding 부분만 담긴 reference를 사용할 수 있다.
Usage
kallisto index -i [output filename 지정] [참조전사체(FASTA-files) 경로]Step2: Quantification
- 한번에 한 sample 씩 (모든 sample 한번에 돌리면 안됨)
Usage:
# paired-end
kallisto quant -i [indexing된 참조전사체 경로] -o [output file이 들어갈 directory 경로] [pairA_1.fq 경로] [pariA_2.fq 경로] -t [thread 수]
# single-end
kallisto quant -i [indexing된 참조전사체 경로] -o [output file이 들어갈 directory 경로] --single -l [Estimated average fragment length(200)] -s [Estimated standard deviation of fragment length(20)] [file_1.fq 경로] -t [thread 수]
Step3: Importing transcript abundance with tximport
- 터미널에서 scp 이용하여 서버에서 로컬로 data 다운받기 (Mac을 사용하는 경우)
scp kjb9412@168.188.119.230:[서버에서 kallisto quant output file 경로] [로컬에 다운받을 경로(/Users/jibeomko/Desktop/~)]- R에서 tximport를 이용하기 위해 tx2gene(id)을 만들고 kallisto output 파일 경로를 생성한다.
- file.path()함수는 주어진 문자열을 이어서 파일에 대한 경로를 만드는 함수이다.
# tx2gene 만들기: 서버의 참조전사체(transcriptome)에서 원하는 id 가져옴
# In linux(server)
grep “>” [참조전사체 파일 경로] > [TXNAME] # >로 시작하는 행 뽑아 TXNAME에 저장 (TXNAME은 transcript id를 담았다)
awk '{sub(/^./,"")}1' [TXNAME] > [TXNAME] # 각 행의 첫번째 문자 제거
awk -F “|” ‘{print $1}’ [TXNAME] > [TXNAME] # |로 필드를 구분하고, 첫번째 필드 출력
grep “>” [참조전사체 파일 경로] > [SYMBOL] # >로 시작하는 행 뽑아 SYMBOL에 저장 (SYMBOL은 gene symbol을 담았다)
awk '{sub(/^./,"")}1' [SYMBOL] > [SYMBOL] # 각 행의 첫번째 문자 제거
awk -F “|” ‘{print $6}’ [SYMBOL] > [SYMBOL] # |로 필드를 구분하고, 여섯번째 필드 출력
# 서버에서 로컬로 TXNAME, SYMBOL 다운받기
# In R
TXNAME <- read.delim('파일 경로', header=F) # TXNAME vector 형태로 loading
SYMBOL <- read.delim('파일 경로', header=F) # SYMBOL vector 형태로 loading
tx2gene <- data.frame(TXNAME, SYMBOL) # tx2gene dataframe 생성
tx2gene <- rename(tx2gene, TXNAME = V1, SYMBOL = V1.1) # tx2gene column name 설정 # kallisto output 파일 이름 지정(순서대로)
sample <- c(’Naive1.tsv’ , ‘Naive2.tsv’ , ‘Naive3.tsv’ , ‘Primary1.tsv’ , ‘Primary2.tsv’ , ‘Primary3.tsv')
# 파일 이름 dataframe 구조로 바꾸기
sample <- as.data.frame(sample)
# file.path()를 이용한 파일 경로 만들기
files <- file.path(‘/Users’ , ‘jibeomko’ , ‘Desktop’, ‘SOX17_RNAseq_Data’, sample$sample)
names(files) <- c(’Naive1.tsv’ , ‘Naive2.tsv’ , ‘Naive3.tsv’ , ‘Primary1.tsv’ , ‘Primary2.tsv’ , ‘Primary3.tsv’) # tximport로 transcript abundance 불러오기
library(tximport)
txi.kallisto.tsv <- tximport(files, type = 'kallisto', tx2gene = tx2gene, ignoreAfterBar = TRUE)
head(txi.kallisto.tsv$counts)Step4: Creating a DESeqDataSet for use with DESeq2
- DESeq2를 활용하기 위한 data set 만들기
library(DESeq2)
sampleTable <- data.frame(condition=factor(rep(c("Naive","Primary"), each = 3))) # metadata 만들기 (sampleTable)
rownames(sampleTable) <- colnames(txi.kallisto.tsv$counts) # metadata의 행이름을 txi.kallisto.tsv의 counts열이름으로 설정
dds <- DESeqDataSetFromTximport(txi.kallisto.tsv, sampleTable, ~condition) # Tximport로부터 불러온 data로 DEG 분석하고, metadata는 sampleTable로 사용, metadata 중 condition을 기준으로 DEG 분석한다.
Step5: DEG analysis using DESeq2
deseq2.dds <- DESeq(dds) # DEG 분석 내용을 deseq2.dds에 할당
deseq2.res <- results(deseq2.dds) # DEG 분석 결과를 deseq2.res에 할당
deseq2.res <- deseq2.res[order(rownames(deseq2.res)),] # DEG 분석 결과를 유전자 이름 순으로 정렬
str(deseq2.res) # 분석 결과 확인
# adjusted p-value가 0.05보다 낮은 경우 확인
dim(deseq2.res[!is.na(deseq2.res$padj) & deseq2.res$padj <= 0.05,]) ## PCA plot 그리기
vsd <- vst(deseq2.dds, blind = F) # 분산 분석
plotPCA(vsd, intgroup = 'condition')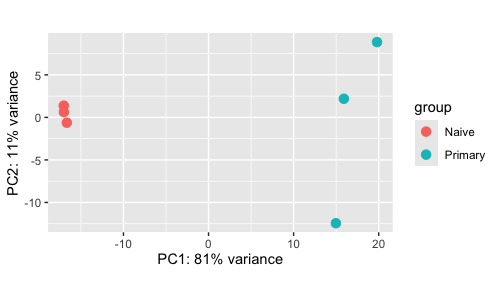
## Heatmap plot 그리기
library(pheatmap)
# Select top 50 DEGs (p-value를 기준으로)
res <- deseq2.res # 새로운 변수 res에 DESeq2 결과 할당
res <- na.omit(res) # NA 제거
res_ordered <- res[order(res$padj),] # p-value 낮은 순으로 정렬해서 새로운 변수에 할당
top_genes <- row.names(res_ordered)[1:50] # p-value 가장 낮은 50개 DEGs top_genes에 할당
# Extract counts and normalize
counts <- counts(deseq2.dds, normalized = T) # 정규화(normalized)된 유전자 발현(count) 데이터(TPM) 추출
counts_top <- counts[top_genes,] # top_genes의 count 데이터(TPM)를 counts_top에 할당
# Log-transform counts
log_counts_top <- log2(counts_top + 1)
# Generate heatmap
pheatmap(log_counts_top, scale = 'row')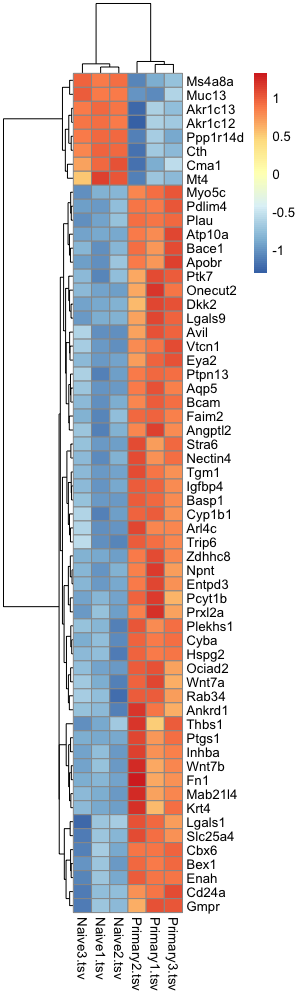
## Main plot 그리기
library(ggplot2)
tmp <- deseq2.res # 새로운 변수 tmp에 DESeq2 결과 할당
tmp <- na.omit(tmp) # NA 제거
plot(tmp$baseMean, tmp$log2FoldChange, pch=20, cex=0.45, ylim=c(-3, 3), log="x", col="darkgray", main="DEG Dessication (pval <= 0.05)", xlab="mean of normalized counts", ylab="Log2 Fold Change")
# p-value가 0.05이하인 부분 빨간점으로 표시
tmp.sig <- tmp[!is.na(tmp$padj) & tmp$padj <= 0.05, ] # tmp의 p-value 중 NA 아니고 0.05이하인 부분을 tmp.sig에 할당
points(tmp.sig$baseMean, tmp.sig$log2FoldChange, pch=20, cex=0.45, col="red") # tmp.sig 빨간점 표시
# 2 FC lines (Fold Change 2, -2 파란선 표시)
abline(h=c(-1,1), col="blue"))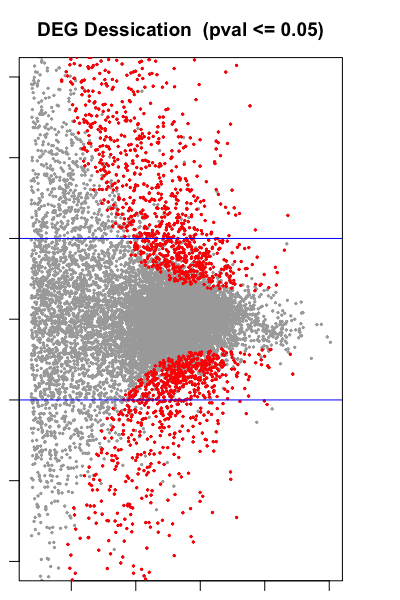
## Volcano plot 그리기
library(ggrepel)
# Prepare the data for plotting
res_df <- as.data.frame(deseq2.res)
res_df <- na.omit(res_df)
res_df$gene <- row.names(res_df)
res_df$gene_symbol <- row.names(res_df)
# Create volcano plot
res_df$gene <- "No"
res_df$gene[res_df$log2FoldChange > 2 & res_df$padj <= 0.05] <- "UP"
res_df$gene[res_df$log2FoldChange < -2 & res_df$padj <= 0.05] <- "DOWN"
mycolors <- c("blue","red","grey")
names(mycolors) <- c("DOWN","UP","NO")
res_df$delabel <- NA
res_df$delabel[res_df$gene != "NO"] <- res_df$gene_symbol[res_df$gene != "NO"]
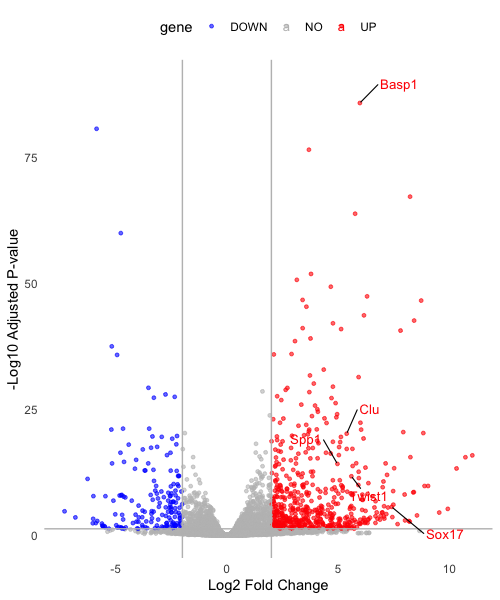
annotation <- res_df %>% filter(gene_symbol %in% c("Sox17","Basp1","Clu","Twist1","Spp1","Igfbp4","S100a4","Ly6d"))
volcano_plot <- ggplot(res_df, aes(x =log2FoldChange, y = -log10(padj),col = gene, label = delabel)) + geom_point(alpha = 0.6, size = 1) + scale_color_manual(values = mycolors) + labs(x = "Log2 Fold Change", y = "-Log10 Adjusted P-value") + theme_minimal() + theme(panel.grid.major = element_blank(), panel.grid.minor = element_blank(), legend.position = "top") + geom_vline(xintercept = c(-2, 2), col = "grey") + geom_hline(yintercept = -log10(0.05), col = "grey") + geom_text_repel(data = annotation, box.padding = 1, segment.color = 'black', segment.size = 0.4, size = 3.5,) + ylim(c(0,90))
print(volcano_plot)