Step 1 : Load required packages
library(dplyr)
library(clusterProfiler)
library(pathview)
library(wordcloud)
library(enrichplot)
library(ggpubr)
library(org.Mm.eg.db)
Step 2 : Import DESeq2 results
res <- deseq2.res
res <- na.omit(res)
head(res)Step 3 : Prepare input
original_gene_list <- res$log2FoldChange # res에서 log2FoldChange 부분 새로운 변수에 할당
names(original_gene_list) <- rownames(res) # 변수 riginal_gene_list에 이름 붙이기
gene_list <- na.omit(original_gene_list) # NA 제거
gene_list = sort(gene_list, decreasing = TRUE) # log2FoldChange 값을 기준으로 내림차순 정렬 (required for clusterProfiler)
sig_genes_df <- subset(res, padj < 0.05) # 통계상 유의미한 값 추출 (pvalue < 0.05 or padj < 0.05)
genes <- sig_genes_df$log2FoldChange # sig_genes_df에서 log2FoldChange 값 추출
names(genes) <- rownames(sig_genes_df) # Name the vector
genes <- na.omit(genes) # NA 제거
genes <- names(genes)[abs(genes) > 2] # Filtering genes with log2fold > 2 / abs()함수는 절대값을 반환함Step 4 : Create enrichGO object
keytypes(org.Mm.eg.db) # org.Mm.eg.db의 keytype 확인
go_enrich <- enrichGO(gene = genes, universe = names(gene_list), OrgDb = org.Mm.eg.db, keyType = "SYMBOL", readable = TRUE, ont = "ALL", pvalueCutoff = 0.01, qvalueCutoff = 0.001)
go_enrichStep 5 : EnrichGO filtering
go_enrich_filtered <- go_enrich
go_enrich_filtered_results <- go_enrich_filtered@result
# Sort GO terms by counting results
go_enrich_filtered_results <- go_enrich_filtered_results %>% arrange(p.adjust)
go_enrich_filtered@result <- go_enrich_filtered_results
# Extract genes for cleaning view
core_genes <- str_split(as.data.frame(go_enrich_filtered_results)[,"geneID"], "/")
# Transform list into matrix - conserving proper list characteristics
core_genes_top <- stringi::stri_list2matrix(core_genes)
core_genes_top <- core_genes_top[1:10, ]
# Re-transforming list
core_genes_top <- as.list(as.data.frame(core_genes_top))
# putting new gene list into go_enrich again -> This is an important process for processing cnetplot
filtered_core_genes_top <- sapply(lapply(core_genes_top, function(x) x[x %in% genes]), paste, collapse = "/")
go_enrich_filtered@result$geneID <- filtered_core_genes_top
Step 6 : Making graphs
## Upset plot
# filtering 안한 data 이용
upsetplot(go_enrich, n = 9, layout = "mds") + ggtitle("Upset-plot with raw GO enrichment data")
# filtered data 이용
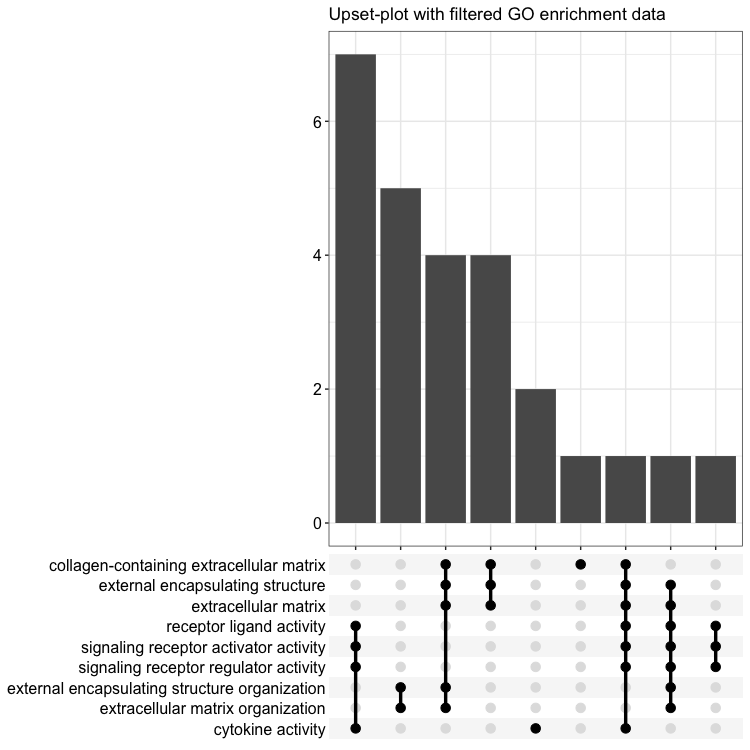
upsetplot(go_enrich_filtered, n = 9, layout = "mds") + ggtitle("Upset-plot with filtered GO enrichment data")
## Bar plot
# filtering 안한 data 이용
barplot(go_enrich, drop = TRUE, showCategory = 15, title = "GO Biological Pathways", font.size = 8) + facet_grid(ONTOLOGY ~ ., scale = "free") + ggtitle("Bar-plot with raw GO enrichment data")
# filtered data 이용
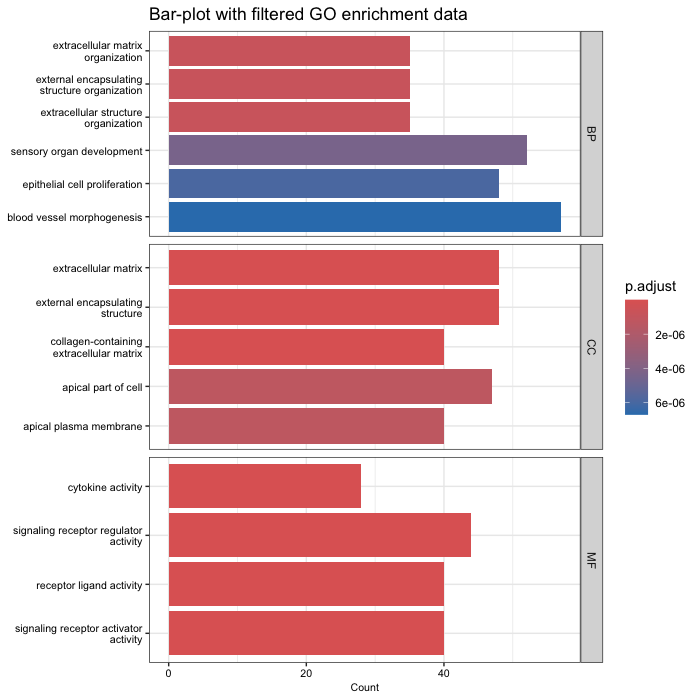
barplot(go_enrich_filtered, drop = TRUE, showCategory = 15, title = "GO Biological Pathways", font.size = 8) + facet_grid(ONTOLOGY ~ ., scale = "free") + ggtitle("Bar-plot with filtered GO enrichment data")
## Dot plot
# filtering 안한 data 이용
dotplot(go_enrich, showCategory = 10) + ggtitle("dotplot for ORA") + facet_grid(ONTOLOGY ~ ., scale = "free") + ggtitle("Dot-plot with raw GO enrichment data")
# filtered data 이용
dotplot(go_enrich_filtered, showCategory = 10) + ggtitle("dotplot for ORA") + facet_grid(ONTOLOGY ~ ., scale = "free") + ggtitle("Dot-plot with filtered GO enrichment data")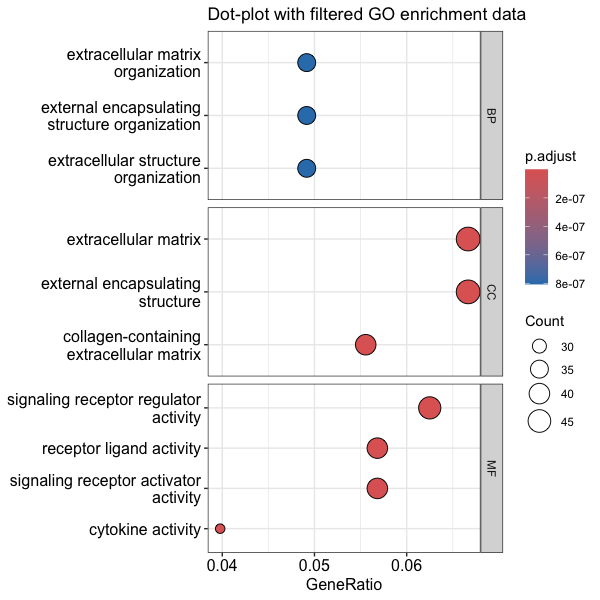
## Enrichment map plot
# filtering 안한 data 이용
mdf <- pairwise_termsim(go_enrich)
emapplot(emdf, showCategory = 15) + ggtitle("Enrichment-map with filtered GO enrichment data")
# filtered data 이용
emdf <- pairwise_termsim(go_enrich_filtered)
emapplot(emdf, showCategory = 15) + ggtitle("Enrichmend-map with raw GO enrichment data")
# plot title 제거
emapplot(emdf, showCategory = 15)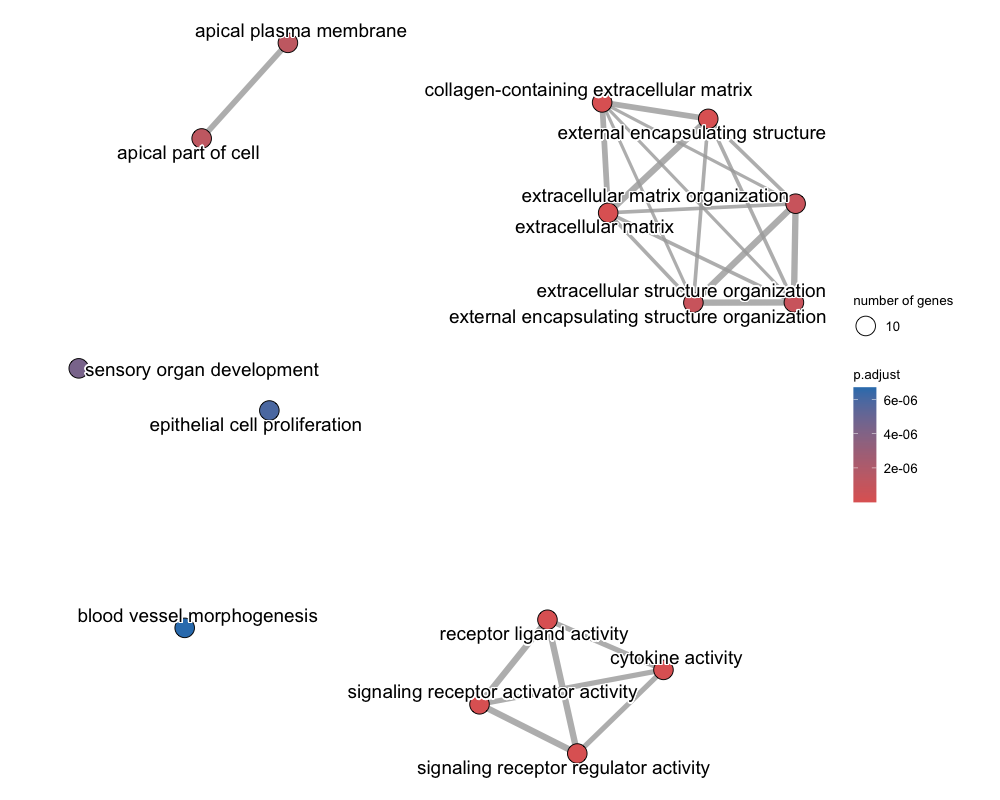
## Category Net plot
# filtering 안한 data 이용
cnetplot(go_enrich, categorySize = "pvalue", foldChange = gene_list) + ggtitle("Standard cnet-plot with raw GO enrichment data")
# filtered data 이용
cnetplot(go_enrich_filtered, categorySize = "pvalue", foldChange = gene_list) + ggtitle("Standard cnet-plot with filtered GO enrichment data")
# plot title 제거
cnetplot(go_enrich_filtered, categorySize = "pvalue", foldChange = gene_list)
# circular 형태로 그리기
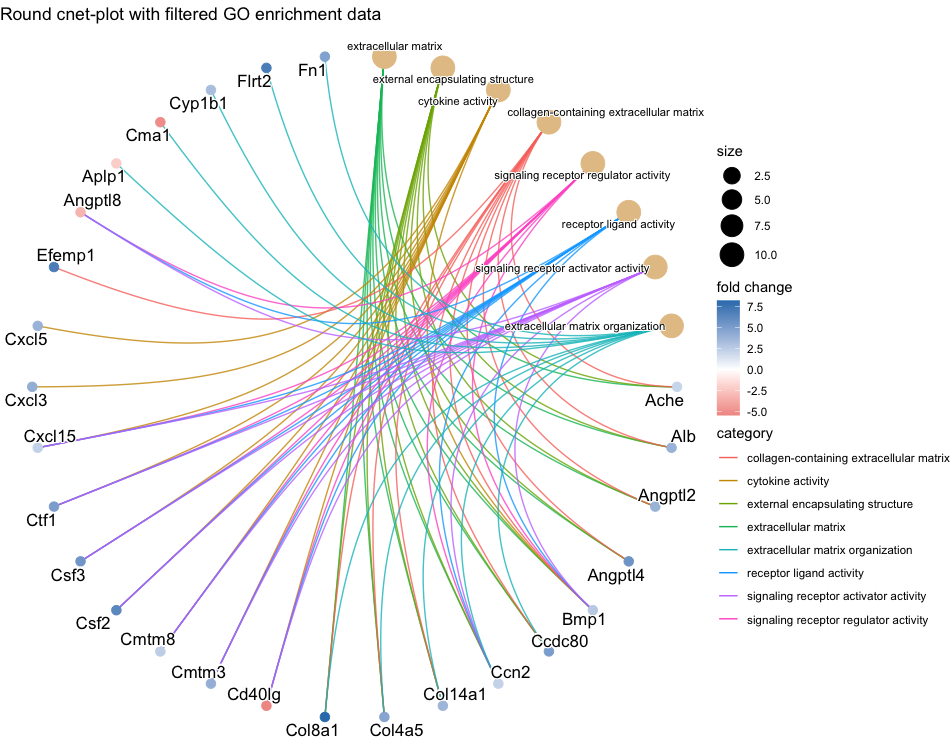
cnetplot(go_enrich_filtered, circular = TRUE, color.params = list(foldChange = gene_list), colorEdge = TRUE, showCategory = 8, cex.params = list(gene_node = 0.01), node_label = "all", cex_label_category = 0.6, cex_label_gene = 0.9) + ggtitle("Round cnet-plot with filtered GO enrichment data")