Step 1 : Load required packages
library(clusterProfiler)
library(msigdbr)
library(org.Mm.eg.db)
library(magrittr)
Step 2 : Import DESeq2 results
deseq2.dds <- DESeq(dds)
deseq2.res <- results(deseq2.dds)
res <- deseq.res
dge_mapped_df <- data.frame(res)Step 3 : Prepare input
# 사용하는 데이터의 종에 따라 species를 바꿔줌
Mm_hallmark_sets <- msigdbr(species = "Mus musculus", category = "H")
# DESeq2 결과 데이터의 중복값이 없는지 확인 => 중복있으면 추가적인 처리과정 필요
any(duplicated(rownames(dge_mapped_df)))
# log2 fold change 값 추출해서 이름붙임
lfc_vector <- dge_mapped_df$log2FoldChange
names(lfc_vector) <- rownames(dge_mapped_df)
# 내림차순 정리
lfc_vector <- sort(lfc_vector, decreasing = TRUE)
head(lfc_vector)Step 4 : Running GSEA
# Set the seed so our results are reproducible:
set.seed(2020)
gsea_results <- GSEA(geneList = lfc_vector, minGSSize = 25, maxGSSize = 500, pvalueCutoff = 0.05, eps = 0, seed = TRUE, pAdjustMethod = "BH", TERM2GENE = dplyr::select(Mm_hallmark_sets, gs_name, gene_symbol))Step 5 : Making graphs
## gseaplot
gsea_result_df <- data.frame(gsea_results@result)
# This returns the 3 rows with the largest NES values
gsea_result_df %>% dplyr::slice_max(NES, n = 3)
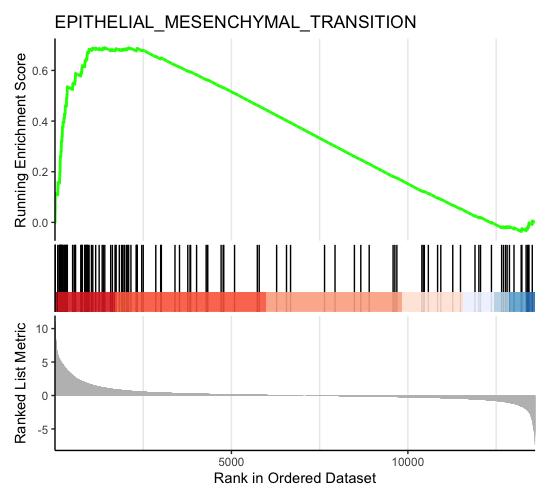
most_positive_nes_plot <- enrichplot::gseaplot(gsea_results, geneSetID = "HALLMARK_EPITHELIAL_MESENCHYMAL_TRANSITION", title = "EPITHELIAL_MESENCHYMAL_TRANSITION", color.line = "#FFCE30")
most_positive_nes_plot
# This returns the 3 rows with the lowest NES values
gsea_result_df %>%dplyr::slice_min(NES, n = 3)
most_negative_nes_plot <- enrichplot::gseaplot(gsea_results, geneSetID = "HALLMARK_UV_RESPONSE_DN", title = "UV_RESPONSE_DN", color.line = "#288BA8")
most_negative_nes_plot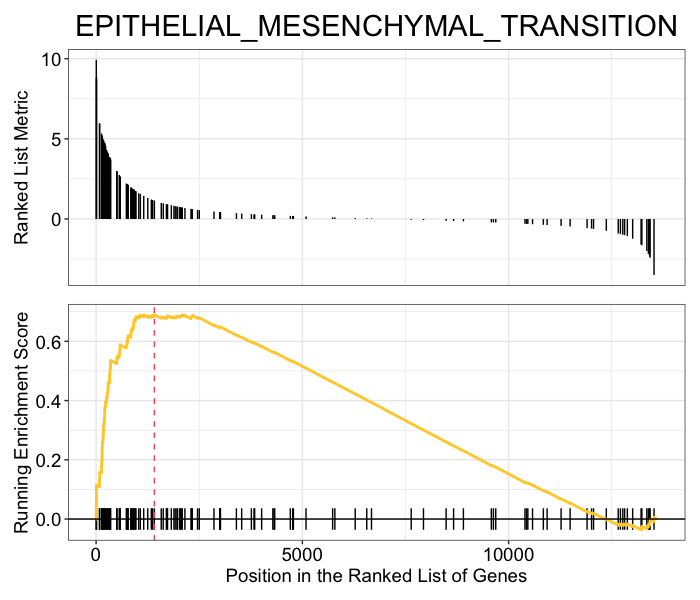
## Ridgeline plot
ridgeplot(gsea_results)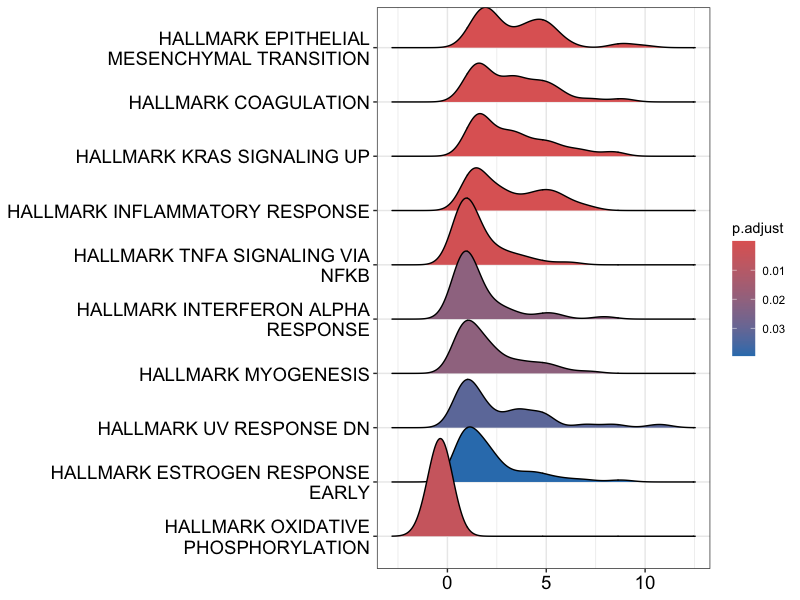
## gseaplot2
gseaplot2(gsea_results, geneSetID = "HALLMARK_EPITHELIAL_MESENCHYMAL_TRANSITION", title = "EPITHELIAL_MESENCHYMAL_TRANSITION")
gseaplot2(gsea_results, geneSetID = 1:3)
gseaplot2(gsea_results, geneSetID = 1:3, pvalue_table = TRUE, color = c("#E495A5", "#86B875", "#7DB0DD"), ES_geom = "dot")