
서론
이번학기에 흥미를 갖고 고른 인공지능과 의료공학 라는 과목 첫 과제입니다.
정말 처음부터 차근차근 시작할 예정이기 때문에, 사전 지식이 있는 분들은
다른 블로그를 찾아 보시는 것을 추천 드립니다.
제가 생명과는 아니라서 생명에 관련된 지식은 스킵하고 핵심적인 부분만 말하자면
1.인간의 몸은 수많은 단백질로 이루어져 있고, 이걸 20개의 아미노산으로 만든다.
2.근데 이게 3D 구조에 종류가 너무 많아 도저히 예측이 안된다.

출처:youtube Kurzgesagt (참고로 여기 재밌습니다. 영어 공부할 때 도움 아주 많이됨)
근데 이거 결국 패턴 분석 아니야? 딥러닝으로 때려박으면 되지 않을까?
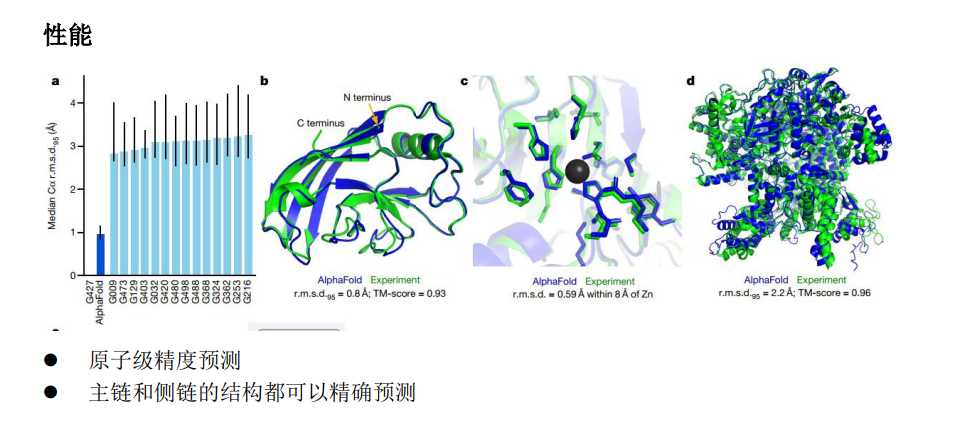
되네?
그렇게 구글 딥마인드가 개발한 alphaFold2 입니다.
처음부터 알파폴드 구조를 이해 하는것은 현재 제 수준에선 불가능한 것 같아, 우선
ProteinStructurePrediction 의 구조 분석부터 시작 해보겠습니다.
ProteinStructurePrediction
- 이 레포지토리는 단백질 구조 예측에 관련된 파이썬 코드를 포함하고 있습니다.
- 주요 파일:
- main.py: 메인 실행 파일
- model.py: 모델 정의 파일
- trainer.py: 훈련 관련 코드
- visualize.py: 결과 시각화 코드
- attention_layer.py: 어텐션 레이어 정의
- helper.py: 도우미 함수
- config.py: 설정 파일
- 라이선스: MIT License
그 외에 사용되는 데이터셋으로 SidechainNet이 있습니다.
SidechainNet에 대한 설명은 다음과 같습니다.
SidechainNet
SidechainNet은 단백질 구조 예측 데이터셋입니다.
이 데이터셋은 Mohammed AlQuraishi에 의해 확장된 ProteinNet1을 직접 확장합니다. SidechainNet은 단백질의 전체 원자 구조(수소를 제외한 백본과 사이드 체인)를 설명하는 단백질 각도와 좌표의 측정값을 추가합니다.
이는 단백질 백본만을 설명하는 ProteinNet과는 대조적입니다.
SidechainNet의 주요 특징:
- 단백질 각도와 좌표의 측정값을 추가하여 전체 원자 단백질 구조를 설명합니다.
- 피클된 Python 사전으로 저장된 SidechainNet 데이터셋을 제공합니다.
- PyTorch에서 SidechainNet 데이터를 효율적으로 로딩하고 배치하는 메서드를 제공합니다.
- 모델 예측에서 단백질 구조 시각화(.pdb, 3Dmol, .gltf)를 생성하는 메서드를 제공합니다.
- 새로운 단백질을 포함하고 데이터셋 조직을 지정하여 SidechainNet을 확장하는 메서드를 제공합니다.
데이터 구조:
- SidechainNet은 기본적으로 Python 사전으로 저장되며, 훈련, 검증, 테스트 분할로 구성됩니다.
- 각 분할 내에서 SidechainNet은 데이터 항목 유형(seq, ang 등)을 키로 하고, 각 유형의 데이터를 포함하는 리스트를 값으로 하는 사전을 매핑합니다.
- 예를 들어,
data = {"train": {"seq": [seq1, seq2, ...], "ang": [ang1, ang2, ...], ...}, "valid-10": {...}, ...}와 같이 구성됩니다.
사용 방법:
- SidechainNet은 Python 사전 또는 PyTorch DataLoaders로 로드할 수 있습니다.
import sidechainnet as scn과data = scn.load(casp_version=12, thinning=30)를 사용하여 SidechainNet을 로드할 수 있습니다.
ProteinStructurePrediction의 모델
ProteinNet이라는 모델을 사용합니다.- 이 모델은 어텐션 메커니즘을 포함하며, 단백질 시퀀스를 입력으로 받아 각 아미노산의 각도를 예측합니다.
- 입력 시퀀스는 정수로 인코딩되며, 이를 임베딩하여 어텐션 레이어와 다른 레이어를 통해 전달합니다.
- 최종 출력은 각 아미노산의 각도에 대한 예측값입니다.
- 즉 ProteinStructurePrediction의 모델은 어텐션 메커니즘을 기반으로 한 기본적인 단백질 구조 예측 모델입니다.
우선 이번 프로젝트에서 가장 중요한 부분은 다음과 같이 요약할 수 있습니다.
- 단백질 구조를 분석할 때 쓰이는 데이터셋(sidechainnet)은 어떠한 형식으로 이루어져 있는가
- prtein_structure_prediction은 어떠한 구조를 가지고 있는가
이중 visualization 함수도 시각화를 위해 아주 중요하지만, 우선 정확도를 높이기 위한 모델의 분석이 먼저라 할 수 있습니다.
이중 Attention 구조 기반인 모델인 attention_layer.py 파일에는 Attention이라는 클래스가 정의되어 있습니다. 이 클래스는 다음 특징들을 갖고 있습니다:
-
초기화 (Constructor)
dim: 입력 차원seq_len: 시퀀스 길이 (기본값: None)heads: 어텐션 헤드의 수 (기본값: 4)dim_head: 각 헤드의 차원 (기본값: 64)dropout: 드롭아웃 비율 (기본값: 0.0)gating: 게이팅 활성화 여부 (기본값: True)- 초기화 과정에서는
to_q,to_kv,to_out,gating에 대한 선형 변환을 정의합니다.
-
Forward Method
forward메소드는 입력x와 다른 선택적 매개변수들을 받아 어텐션 메커니즘을 계산합니다.q,k,v는 각각 query, key, value를 나타내며, 이들은 입력x와context를 통해 계산됩니다.- 어텐션 점수(
dots)는q와k의 유사도를 기반으로 계산됩니다. - 어텐션 점수에 마스크를 적용하고, softmax를 통해 정규화합니다.
- 어텐션 가중치를 사용하여
v를 가중 평균합니다. - 게이팅 메커니즘을 통해 출력을 조절합니다.
- 최종 출력은
to_out선형 변환을 통해 반환됩니다.
이 클래스는 멀티-헤드 어텐션 메커니즘을 구현하며, 게이팅 메커니즘을 통해 출력을 추가적으로 조절하며,
게이팅 메커니즘은 출력에 대한 정보 흐름을 조절하여 모델의 성능을 향상시킬 수 있습니다.
여기서 Attention Mechanism 에 대하여 조금 더 자세히 알아봅시다.
어텐션 메커니즘을 자세히 이해하기 위해선 seq2seq, transformer 모델을 분석 해보는 것이 좋을 것 같습니다.
우선 가장 핵심적인 사전 지식인 인코더(encoder) 와 디코더(decoder) 에 대해 알아봅시다.
우선 Seq2Seq(Sequence-to-Sequence) 모델과 Encoder-Decoder 구조는 매우 밀접한 관련이 있으며, Seq2Seq 모델이 Encoder-Decoder 구조를 기반으로 합니다.
그러나 두 용어는 다음과 같이 약간 다르게 사용될 수 있습니다:
Seq2Seq (Sequence-to-Sequence) Model:
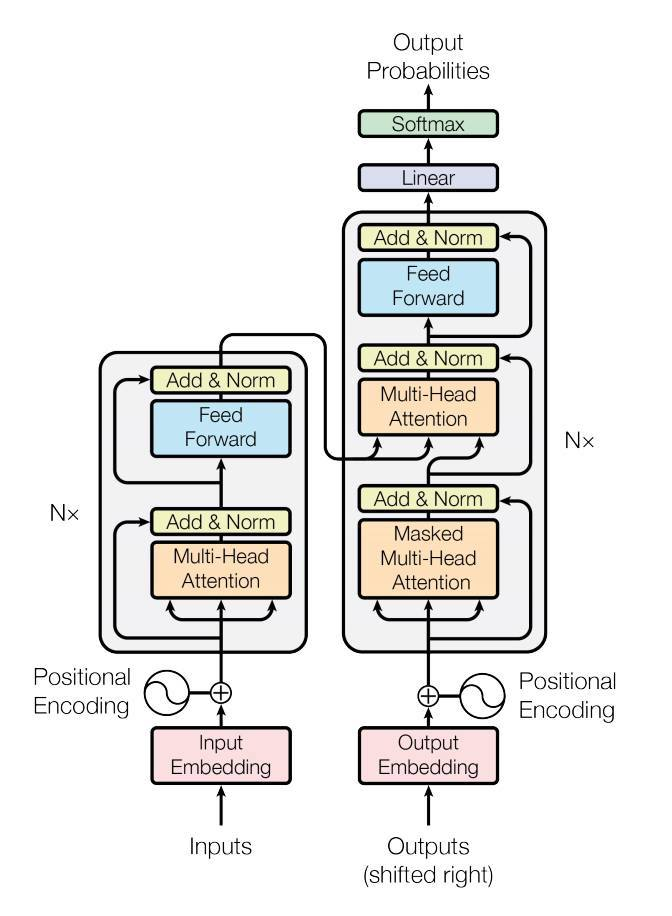
- 정의: Seq2Seq 모델은 입력 시퀀스를 출력 시퀀스로 변환하는 모델입니다. 이 모델은 주로 기계 번역, 질문 응답, 텍스트 요약 등의 작업에 사용됩니다.
- 구조: Seq2Seq 모델은 기본적으로 Encoder와 Decoder로 구성되어 있습니다.
Encoder는 입력 시퀀스를 고정 길이의 벡터로 인코딩하고, Decoder는 이 벡터를 사용하여 출력 시퀀스를 생성합니다. - 특징: Seq2Seq 모델은 RNN, LSTM, GRU 등의 순환 신경망을 사용할 수 있으며, Attention 메커니즘과 함께 사용되기도 합니다.
Encoder Decoder 구조:
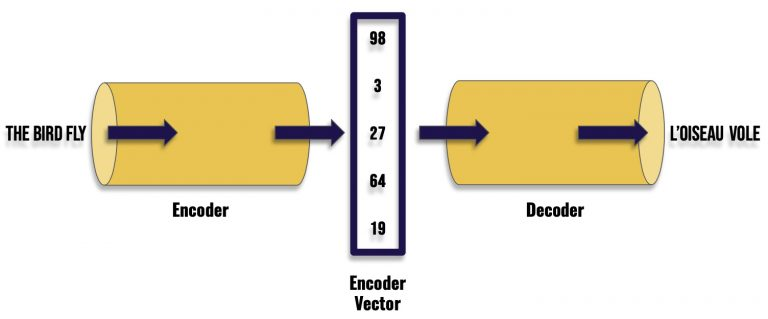
Encoder:
- 역할: Encoder는 입력 데이터(예: 텍스트, 이미지)를 고차원에서 저차원으로 변환합니다. 이 과정에서 입력 데이터의 중요한 특징이나 패턴을 추출합니다.
- 작동 방식: Encoder는 입력 데이터를 받아 내부 상태를 업데이트하며, 이 상태는 입력 데이터의 압축된 표현입니다. 이 상태는 "context" 또는 "encoding"이라고도 합니다.
- 구조: Encoder는 일반적으로 RNN, LSTM, GRU, Transformer 등의 신경망 구조를 사용합니다.
Decoder:
- 역할: Decoder는 Encoder가 생성한 저차원 데이터를 다시 고차원 데이터로 변환합니다. 이 과정에서 원하는 출력 데이터(예: 텍스트, 이미지)가 생성됩니다.
- 작동 방식: Decoder는 Encoder의 출력을 입력으로 받아, 이를 바탕으로 원하는 출력 데이터를 생성합니다. 이 과정에서 Decoder는 내부 상태를 업데이트합니다.
- 구조: Decoder 역시 Encoder와 유사한 신경망 구조를 사용하며, 종종 Encoder와 대칭적인 구조를 가집니다.
Encoder-Decoder 구조의 활용:
- 기계 번역: Encoder는 원문 텍스트를 인코딩하고, Decoder는 이를 바탕으로 대상 언어의 텍스트를 생성합니다.
- 이미지 캡셔닝: Encoder는 이미지를 인코딩하고, Decoder는 이를 바탕으로 이미지에 대한 설명 텍스트를 생성합니다.
- 음성 인식: Encoder는 음성 신호를 인코딩하고, Decoder는 이를 바탕으로 텍스트로 변환합니다.
예시:
-
Encoder가 "prendre une expression au pied de la lettre"라는 문장을 인코딩하면,
이를 바탕으로 Decoder는 "take an expression literally"라는 문장을 생성합니다.
이를 통해 Encoder-Decoder 구조가 문장의 전체적인 의미를 파악하여 번역하는 것을 확인할 수 있습니다
(출처). -
이는 다음과 같이 요약할 수 있습니다.
-
Seq2Seq 모델은 Encoder-Decoder 구조를 사용하여 시퀀스 데이터를 처리하는 특정 유형의 모델을 지칭합니다.
-
Encoder-Decoder 구조는 더 일반적인 개념으로, Seq2Seq 모델 외에도 다양한 딥러닝 모델의 구조를 설명하는 데 사용됩니다.
그래서 왜 Attenion은 설명 안하고 언어 모델을 설명하고 있냐고 하면, 어텐션 메커니즘의 기본 컨셉이 encoder-decoder 에 기반하기 때문입니다.
또한 단백질 구조도 아미노산으로 구성된 "언어" 라고 할 수 있기 때문에 다음 컨셉이 쓰인것이 아닐까 추측합니다.
Attention 은 어째서 등장했는가?
seq2seq 모델은 인코더에서 입력 시퀀스를 컨텍스트 벡터라는 하나의 고정된 크기의 벡터 표현으로 압축하고, 디코더는 이 컨텍스트 벡터를 통해서 출력 시퀀스를 만들어냅니다.
하지만 이러한 RNN에 기반한 seq2seq 모델에는 크게 두 가지 문제가 있습니다.
1. 하나의 고정된 크기의 벡터에 모든 정보를 압축하려고 하니까 정보 손실이 발생합니다.
2. RNN의 고질적인 문제인 기울기 소실(vanishing gradient) 문제가 존재합니다.
결국 이는 기계 번역 분야에서 입력 문장이 길면 번역 품질이 떨어지는 현상으로 나타났습니다.
단백질은 20개의 아미노산으로 우주에 존재하는 모든 별보다 많은 경우의 수를 만들 수 있습니다.
alphaFold2가 등장하기 전에 단백질 구조의 분석이 힘들고 천문학적 비용을 소모한 이유입니다.
이를 위한 대안으로 입력 시퀀스가 길어지면 출력 시퀀스의 정확도가 떨어지는 것을 보정해주기 위한 등장한 기법인 어텐션(attention)에 대해 알아봅시다.
어텐션 메커니즘은 딥 러닝 모델, 특히 시퀀스-투-시퀀스(Sequence-to-Sequence, Seq2Seq) 모델에서 중요한 부분에 집중할 수 있게 해주는 기술입니다. 어텐션은 입력 시퀀스의 각 요소에 가중치를 할당하여 모델이 중요한 정보에 더 집중할 수 있도록 합니다.
어텐션 메커니즘에 대해 자세히 설명이 되어 있습니다. 이를 우선 확인 해 보는것을
추천 드립니다.
data structure에 나오는 key-value 구조, embedding ,context
에 대한 약간의 지식이 필요합니다. 요약하자면 다음과 같습니다.
어텐션의 Embedding, Query, Key, Value 설명:
"임베딩" 이라는 용어는 주로 자연어 처리(NLP) 및 기계 학습 분야에서 사용되며, 고차원의 데이터를 저차원의 벡터 공간에 표현하는 과정을 의미합니다.
이 과정을 통해 데이터 간의 관계나 유사도를 벡터 공간에서의 거리나 방향으로 표현할 수 있습니다.
임베딩의 주요 개념:
-
단어 임베딩 (Word Embedding):
- 정의: 단어 임베딩은 각 단어를 고정된 크기의 실수 벡터로 변환하는 기술입니다.
- 목적: 텍스트 데이터를 컴퓨터가 이해하고 처리할 수 있는 형태로 변환합니다.
- 예시: "사과"라는 단어를
[0.1, 0.3, -0.2]와 같은 벡터로 표현합니다.
-
문장 임베딩 (Sentence Embedding):
- 정의: 문장 임베딩은 전체 문장을 하나의 벡터로 표현합니다.
- 목적: 문장 간의 유사도를 계산하거나, 문장을 입력으로 사용하는 모델에 적용합니다.
- 예시: "나는 학교에 간다"라는 문장을
[0.5, -0.1, 0.3]와 같은 벡터로 표현합니다.
-
임베딩 레이어 (Embedding Layer):
- 정의: 딥 러닝 모델에서 사용되는 임베딩 레이어는 특정 단어의 인덱스를 해당 단어의 임베딩 벡터로 매핑합니다.
- 목적: 모델의 입력으로 사용되는 단어나 문장을 연속적인 벡터로 변환합니다.
- 예시: 단어 "책"의 인덱스가
3이라면, 임베딩 레이어는 이를[0.2, -0.1, 0.4]와 같은 벡터로 변환합니다.
임베딩의 장점:
- 차원 축소: 고차원의 데이터를 저차원의 벡터로 변환하여, 계산 효율성을 높이고, 데이터의 복잡성을 줄입니다.
- 유사도 표현: 벡터 공간에서의 거리나 방향을 사용하여 데이터 간의 유사도를 측정합니다.
- 희소성 해소: 원-핫 인코딩과 같은 희소 표현을 밀집 벡터로 변환하여, 데이터의 표현력을 향상합니다.
예시:
- 원-핫 인코딩: "사과" =
[1, 0, 0, 0], "바나나" =[0, 1, 0, 0] - 단어 임베딩: "사과" =
[0.1, 0.3, -0.2], "바나나" =[0.2, -0.1, 0.3]
단어 임베딩을 통해 단어나 문장의 의미를 벡터로 효과적으로 표현할 수 있으며, 이를 기반으로 다양한 NLP 작업을 수행할 수 있습니다.
-
Query (Q):
- Query는 현재의 관점이나 참조점을 나타냅니다.
- Decoder의 각 단계에서 생성되며, 어떤 정보를 찾아야 하는지를 표현합니다.
-
Key (K):
- Key는 각 입력 요소의 식별자 또는 주소 역할을 합니다.
- Encoder의 각 입력 요소에 대해 생성되며, Query가 어떤 정보에 접근할 수 있는지를 결정합니다.
-
Value (V):
- Value는 실제 정보의 내용을 나타냅니다.
- Key와 연관된 실제 데이터로, Query가 참조하는 정보의 내용을 표현합니다.
context는 뜻이 여러가지가 있는데 다 알아두면 좋습니다.
1. 프로그래밍 및 컴퓨팅:
정의: 실행 환경이나 상태를 나타내며, 특정 작업이나 함수가 어떻게 동작해야 하는지에 대한 정보를 제공합니다.
예시: 웹 개발에서 HTTP 요청의 "context"는 요청에 대한 메타데이터, 헤더, 본문 등을 포함합니다.
2. 자연어 처리 (NLP):
정의: 단어나 문장의 의미를 이해하거나 해석하는 데 필요한 주변 텍스트나 환경을 의미합니다.
예시: "배"라는 단어가 "나무"와 함께 사용되면 과일을 의미하고, "바다"와 함께 사용되면 선박을 의미합니다. 여기서 "나무"와 "바다"는 "배"의 "context"입니다.
3. 딥 러닝 및 기계 학습:
정의: 모델이 데이터를 해석하거나 예측을 수행하는 데 사용하는 입력 데이터의 일부입니다.
예시: 시퀀스-투-시퀀스 모델에서 "context" 벡터는 인코더의 출력으로, 디코더가 시퀀스를 생성하는 데 사용되는 정보를 포함합니다.
어텐션 메커니즘의 단계별 설명:
-
입력 준비:
- 어텐션 메커니즘은 주로 Encoder-Decoder 구조에서 사용됩니다.
- Encoder는 입력 시퀀스를 고정 길이의 컨텍스트 벡터로 변환합니다.
-
가중치 계산:
- Decoder의 각 시점에서, 입력 시퀀스의 각 요소에 대한 가중치를 계산합니다.
- 가중치는 Decoder의 현재 상태와 Encoder의 각 상태와의 유사도에 기반합니다.
- 유사도를 계산하는 방법은 다양하며, 대표적으로 dot-product attention, scaled dot-product attention, additive attention 등이 있습니다.
-
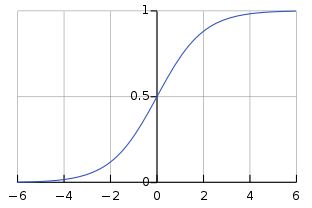
가중치 정규화:
- 계산된 가중치를 정규화하여 합이 1이 되도록 만듭니다. 이를 통해 가중치를 확률 분포로 변환합니다.
- 정규화를 위해 softmax 함수를 사용합니다.
-
가중 평균 계산:
- 정규화된 가중치와 Encoder의 상태를 곱한 후, 이를 합하여 가중 평균을 계산합니다.
- 가중 평균은 입력 시퀀스의 각 요소가 Decoder의 현재 시점에 얼마나 영향을 미치는지를 반영합니다.
-
출력 생성:
- 가중 평균과 Decoder의 현재 상태를 결합하여 출력을 생성합니다.
- 결합 방법은 모델의 구조에 따라 다를 수 있습니다.
예시:
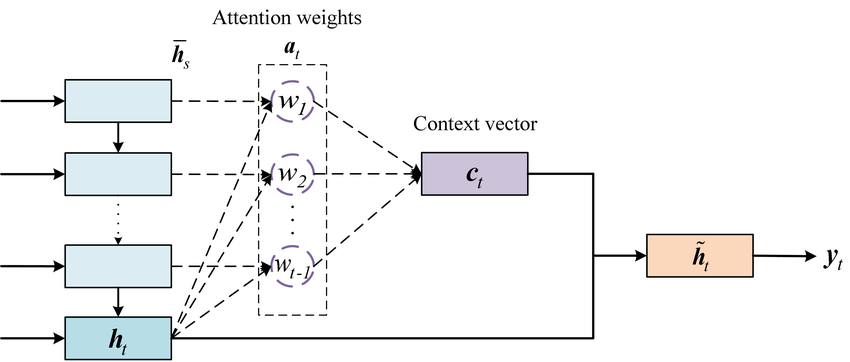
- 입력 시퀀스: "I love deep learning"
- Encoder 상태: [0.1, 0.3, 0.5, 0.7]
- 가중치 계산: Decoder의 현재 상태와 각 Encoder 상태의 유사도를 계산
- 가중치 정규화: softmax([0.1, 0.2, 0.3, 0.4]) = [0.17, 0.24, 0.27, 0.32]
- 가중 평균 계산: (0.17 0.1) + (0.24 0.3) + (0.27 0.5) + (0.32 0.7) = 0.431
- 출력 생성: 가중 평균과 Decoder의 현재 상태를 결합하여 출력 생성
자 이제 어텐션에 대한 기본 개념을 알았으니 단백질 구조 예측 코드에 사용된
멀티 헤드 어텐션에 대해 알아봅시다.
멀티-헤드 어텐션 (Multi-Head Attention):
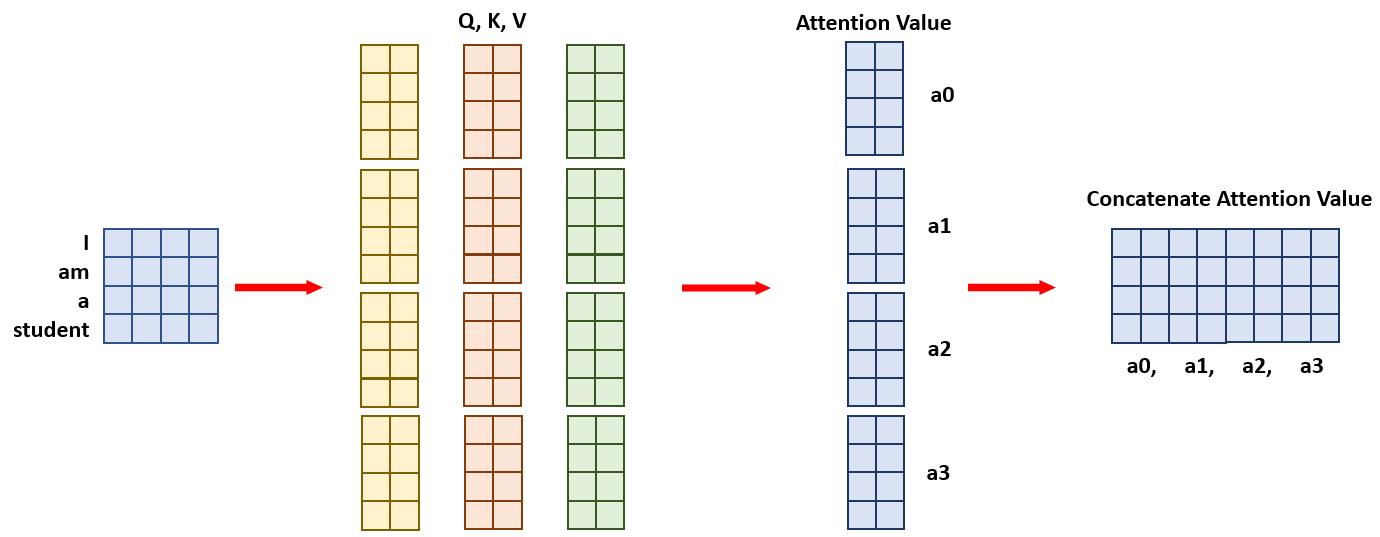
멀티-헤드 어텐션은 어텐션 메커니즘을 여러 번 병렬로 수행하여, 입력 시퀀스의 다양한 부분에 attention 할 수 있게 합니다.
-
단계별 설명:
- 분할: 입력 임베딩을 여러 개의
head로 분할합니다. 각 헤드는 입력의 서로 다른 부분을 처리합니다. - 어텐션 계산: 각 헤드에서 어텐션 가중치를 독립적으로 계산합니다.
- 결합: 각 헤드의 어텐션 출력을 결합하여 최종 출력을 생성합니다.
- 분할: 입력 임베딩을 여러 개의
-
장점:
- 여러 헤드가 서로 다른 어텐션 가중치를 학습하므로, 모델은 입력의 다양한 특징을 동시에 포착할 수 있습니다.
- 이를 통해 모델은 복잡한 패턴과 관계를 더 효과적으로 학습합니다.
예를 들어보겠습니다.
Multi-Head Attention 예제:
- 입력 문장: "나는 딥 러닝을 공부한다"
- 목표 문장: "I study deep learning"
-
입력 준비:
- 입력 문장의 각 단어를 임베딩하여 Key와 Value를 생성합니다.
- 목표 문장의 이전 단어를 임베딩하여 Query를 생성합니다.
-
멀티-헤드 어텐션:
- 입력 임베딩을 여러 헤드로 분할합니다.
- 각 헤드에서 Query, Key, Value를 사용하여 어텐션 가중치를 계산합니다.
- 예를 들어, 첫 번째 헤드는 "나는"과 "공부한다"에 집중하고, 두 번째 헤드는 "딥 러닝을"에 집중할 수 있습니다.
-
가중치 계산:
- 각 헤드에서 계산된 어텐션 가중치를 사용하여 Value의 가중 평균을 계산합니다.
-
결합 및 출력:
- 각 헤드의 가중 평균을 결합하여 최종 출력을 생성합니다.
- 이 출력은 다음 단어를 예측하는 데 사용됩니다.
- "나는 딥 러닝을 공부한다"라는 문장에서, "공부한다"라는 단어에 집중하여 "study"라는 단어를 예측하는 과정에서, 멀티-헤드 어텐션은 문장의 다양한 부분에 집중할 수 있기 때문에, 이를 통해 모델은 문맥을 더 정확하게 이해하고, 정확한 번역을 생성할 수 있습니다.
게이팅 메커니즘 (Gating Mechanism):
게이팅 메커니즘은 신경망의 정보 흐름을 조절하여, 모델이 중요한 정보를 선택적으로 사용하도록 합니다.
-
구성 요소:
- 업데이트 게이트: 현재 정보를 얼마나 사용할지 결정합니다.
- 리셋 게이트: 과거 정보를 얼마나 '잊어버릴지' 결정합니다.
- 메모리 셀: 게이트의 출력과 현재 입력을 사용하여 새로운 정보를 생성합니다.
-
작동 방식:
- 게이트는 0과 1 사이의 값을 가지며, 이 값에 따라 정보의 흐름을 조절합니다.
- 예를 들어, 업데이트 게이트가 0.8이면, 80%의 현재 정보와 20%의 과거 정보를 결합하여 새로운 정보를 생성합니다.
-
활용 분야:
- 게이팅 메커니즘은 주로 순환 신경망(RNN)의 변형인 GRU(Gated Recurrent Unit)나 LSTM(Long Short-Term Memory)에서 사용됩니다.
- 이를 통해 모델은 시퀀스 데이터에서 장기 의존성을 더 효과적으로 학습합니다.
즉 멀티-헤드 어텐션은 입력의 다양한 특징을 동시에 포착하며, 게이팅 메커니즘은 정보 흐름을 세밀하게 조절합니다. 이 두 메커니즘은 딥 러닝 모델이 복잡한 패턴과 관계를 효과적으로 학습하는 데 도움을 줍니다.
구조에 필요한 지식은 어느정도 파악 되었습니다.
이제 코드를 분석해봅시다.
attention_layer.py
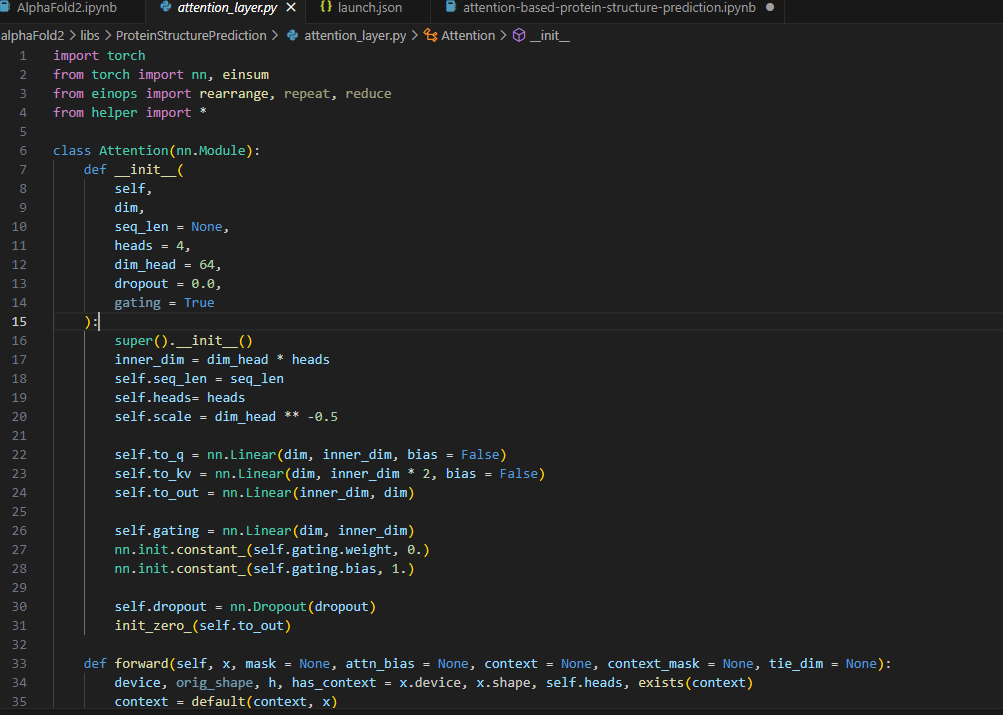
클래스로 이루어져 있고, 다음과 같은 기능들을 가집니다.
-
초기화 (Constructor)
dim: 입력 차원seq_len: 시퀀스 길이 (기본값: None)heads: 어텐션 헤드의 수 (기본값: 4)dim_head: 각 헤드의 차원 (기본값: 64)dropout: 드롭아웃 비율 (기본값: 0.0)gating: 게이팅 활성화 여부 (기본값: True)- 초기화 과정에서는
to_q,to_kv,to_out,gating에 대한 선형 변환을 정의합니다.
-
Forward Method
forward메소드는 입력x와 다른 선택적 매개변수들을 받아 어텐션 메커니즘을 계산합니다.q,k,v는 각각 query, key, value를 나타내며, 이들은 입력x와context를 통해 계산됩니다.- 어텐션 점수(
dots)는q와k의 유사도를 기반으로 계산됩니다. - 어텐션 점수에 마스크를 적용하고, softmax를 통해 정규화합니다.
- 어텐션 가중치를 사용하여
v를 가중 평균합니다. - 게이팅 메커니즘을 통해 출력을 조절합니다.
- 최종 출력은
to_out선형 변환을 통해 반환됩니다.
즉 멀티-헤드 어텐션 메커니즘을 구현하며, 게이팅 메커니즘을 통해 출력을 추가적으로 조절합니다.
그 다음 게이팅 메커니즘으로 출력에 대한 정보 흐름을 조절하여 모델의 성능을 향상시킬 수 있습니다.
config.py
import argparse
def str2bool(v):
return v.lower() in ('true')
def get_parameters():
parser = argparse.ArgumentParser()
# Model Hyper-parameters
parser.add_argument('--d_in', dest='d_in', default=49, type=int,
help="Model input dimension.")
parser.add_argument('--d_hidden', dest='d_hidden', default=512, type=int,
help="Dimensionality of RNN hidden state.")
parser.add_argument('--dim', dest='dim', default=256, type=int,
help="Attention Layer Dim.")
parser.add_argument('--d_embedding', dest='d_embedding', default=32, type=int,
help="Embedding dimension.")
parser.add_argument('--n_heads', dest='n_heads', default=8, type=int,
help="Number of heads in Attention Layer.")
parser.add_argument('-h_dim','--head_dim', dest='head_dim', default=64, type=int,
help="Dimension of heads in Attention Layer.")
parser.add_argument('-int_seq','--integer_sequence', dest='integer_sequence', type=str2bool, default=False,
help="Dimension of heads in Attention Layer.")
# Training parameters
parser.add_argument('-lr', '--learning_rate', dest='learning_rate', default=0.001, type=float,
help="Learning Rate.")
parser.add_argument('-e', '--epoch', type=int, default=10, help="Training Epochs.")
parser.add_argument('-b', '--batch', dest='batch',type=int, default=4,
help="Batch size during each training step.")
parser.add_argument('-t','--train', type=str2bool, default=False,help="True when train the model, \
else used for testing.")
parser.add_argument('--mode', type=str, default='pssms', choices=['pssms', 'seqs'],
help="Mode of trainig the model. Select the input of model either to be PSSM-Position Specific Scoring Matrix \
or Seqs(Protein Sequence)")
# Validation
parser.add_argument('--idx', type=int, default=0,
help="Validation index")
# Base Directory
parser.add_argument('-m', '--model_save_path', type=str, default='./models',
help="Path to Saved model directory.")
return parser.parse_args()
config.py 파일은 모델과 훈련에 필요한 하이퍼파라미터를 설정하는 코드를 포함하고 있습니다. 파일의 주요 내용은 다음과 같습니다:
-
str2bool 함수: 문자열을 Boolean 값으로 변환하는 함수입니다. 'true' 문자열을
True로 변환합니다. -
get_parameters 함수: 이 함수는 모델과 훈련의 하이퍼파라미터를 설정합니다.
d_in: 모델의 입력 차원 (기본값: 49)d_hidden: RNN의 은닉 상태 차원 (기본값: 512)dim: 어텐션 레이어의 차원 (기본값: 256)d_embedding: 임베딩 차원 (기본값: 32)n_heads: 어텐션 레이어의 헤드 수 (기본값: 8)head_dim: 어텐션 레이어의 헤드 차원 (기본값: 64)integer_sequence: 어텐션 레이어의 헤드 차원을 정수 시퀀스로 설정 (기본값: False)learning_rate: 학습률 (기본값: 0.001)epoch: 훈련 에포크 수 (기본값: 10)batch: 배치 크기 (기본값: 4)train: 모델 훈련 여부 (기본값: False)mode: 모델 훈련 모드. 'pssms' 또는 'seqs'를 선택 (기본값: 'pssms')idx: 검증 인덱스 (기본값: 0)model_save_path: 모델 저장 경로 (기본값: './models')
argparse 라이브러리를 사용하여 커맨드 라인에서 하이퍼파라미터를 쉽게 설정할 수 있게 합니다.
model.py
import torch
from torch import nn
from attention_layer import Attention
import sidechainnet as scn
class ProteinNet(nn.Module):
"""A protein sequence-to-angle model that consumes integer-coded sequences."""
def __init__(self,
d_hidden,
dim,
d_in=21,
d_embedding=32,
heads = 8,
dim_head = 64,
integer_sequence=True,
n_angles=scn.structure.build_info.NUM_ANGLES):
super(ProteinNet, self).__init__()
# Dimensionality of RNN hidden state
self.d_hidden = d_hidden
self.attn = Attention(dim = dim,
heads = heads,
dim_head = dim_head)
# Output vector dimensionality (per amino acid)
self.d_out = n_angles * 2
# Output projection layer. (from RNN -> target tensor)
self.hidden2out = nn.Sequential(
nn.Linear(d_embedding, d_hidden),
nn.GELU(),
nn.Linear(d_hidden, self.d_out)
)
self.out2attn = nn.Linear(self.d_out, dim)
self.final = nn.Sequential(
nn.GELU(),
nn.Linear(dim, self.d_out))
self.norm_0 = nn.LayerNorm([dim])
self.norm_1 = nn.LayerNorm([dim])
self.activation_0 = nn.GELU()
self.activation_1 = nn.GELU()
# Activation function for the output values (bounds values to [-1, 1])
self.output_activation = torch.nn.Tanh()
# We embed our model's input differently depending on the type of input
self.integer_sequence = integer_sequence
if self.integer_sequence:
self.input_embedding = torch.nn.Embedding(d_in, d_embedding, padding_idx=20)
else:
self.input_embedding = torch.nn.Linear(d_in, d_embedding)
def get_lengths(self, sequence):
"""Compute the lengths of each sequence in the batch."""
if self.integer_sequence:
lengths = sequence.shape[-1] - (sequence == 20).sum(axis=1)
else:
lengths = sequence.shape[1] - (sequence == 0).all(axis=-1).sum(axis=1)
return lengths.cpu()
def forward(self, sequence, mask=None):
"""Run one forward step of the model."""
# First, we compute sequence lengths
lengths = self.get_lengths(sequence)
# Next, we embed our input tensors for input to the RNN
sequence = self.input_embedding(sequence)
# Then we pass in our data into the RNN via PyTorch's pack_padded_sequences
sequence = torch.nn.utils.rnn.pack_padded_sequence(sequence,
lengths,
batch_first=True,
enforce_sorted=False)
output, output_lengths = torch.nn.utils.rnn.pad_packed_sequence(sequence,
batch_first=True)
# At this point, output has the same dimentionality as the RNN's hidden
# state: i.e. (batch, length, d_hidden).
# We use a linear transformation to transform our output tensor into the
# correct dimensionality (batch, length, 24)
output = self.hidden2out(output)
output = self.out2attn(output)
output = self.activation_0(output)
output = self.norm_0(output)
output = self.attn(output, mask=mask)
output = self.activation_1(output)
output = self.norm_1(output)
output = self.final(output)
# Next, we need to bound the output values between [-1, 1]
output = self.output_activation(output)
# Finally, reshape the output to be (batch, length, angle, (sin/cos val))
output = output.view(output.shape[0], output.shape[1], 12, 2)
return outputmodel.py 에서 ProteinNet이라는 모델을 정의합니다.
이 모델은 단백질 서열을 입력으로 받아 각 아미노산의 각도를 예측하며,
주요 구성 요소는 다음과 같습니다:
ProteinNet클래스는nn.Module을 상속받아 구현됩니다.__init__메서드에서는 모델의 하이퍼파라미터와 레이어를 초기화합니다.Attention클래스를attention_layer.py에서 가져와 사용합니다.forward메서드에서는 입력 서열을 임베딩하고, RNN을 통과시킨 후, 출력을 생성합니다.
config.py 파일은 모델의 하이퍼파라미터와 학습 파라미터를 정의합니다. argparse를 사용하여 커맨드라인에서 파라미터를 입력받을 수 있게 설정합니다.
get_parameters함수에서argparse.ArgumentParser객체를 생성하고, 모델과 학습에 필요한 파라미터를 추가합니다.str2bool함수는 문자열을 bool 값으로 변환합니다.
model.py와 attention_layer.py의 상관 관계:
model.py는attention_layer.py에서 정의된Attention클래스를 사용합니다.ProteinNet모델은Attention레이어를 포함합니다.
즉 config.py는 파라미터를 제공하며, attention_layer.py는 어텐션 메커니즘을 구현하고, model.py는 최종 모델을 정의합니다.
훈련 방법

이 두 방식은 단백질 구조 예측 문제를 해결하기 위한 두 가지 다른 접근 방식을 설명합니다.
각 방식은 모델의 입력 데이터와 목표 출력, 그리고 모델의 구조와 학습 방식에 차이를 보입니다. 각 방식의 주요 차이점은 다음과 같습니다:
1. Training with PSSMs:
- 입력 (Input):
- Sequences (단백질 서열)
- PSSMs (Position Specific Scoring Matrix)
- Secondary Structures (2차 구조)
- Information Content (정보 함량)
- 목표 (Target): 각 아미노산의 각도 (Angles)
- 모델 구조:
- 입력 데이터를 결합하여 하나의 텐서로 만듭니다.
- Hidden state dimension이 1024로 증가합니다.
- PSSMs, secondary structure, information content를 모델 입력에 추가합니다.
- 데이터셋: CASP 12 (30% thinning)
- 학습 방식: PSSMs 방식을 사용하여 학습합니다.
훈련 방법
python main.py --mode pssms --train True \
--epoch 10 --batch 4 --d_in 49 \
--d_hidden 512 --dim 256 \
--d_embedding 32 --n_heads 8 \
--h_dim 64 --integer_sequence False --model_save_path './models'
2. Training with Protein Sequence:
- 입력 (Input): 단백질 서열만 사용합니다.
- 목표 (Target): 각 아미노산의 각도의 sin과 cos 값을 예측합니다.
- 모델 구조:
- Transformer (Attention) 모델을 사용합니다.
- 각도의 순환적 특성을 다루기 위해, 각도의 sin과 cos 값을 예측하고, atan2 함수를 사용하여 각도를 복구합니다.
- 모델 출력은 ( L \times 12 \times 2 )의 형태를 가집니다.
- 학습 방식: 단백질 서열 정보만을 사용하여 학습합니다.
훈련 방법
python main.py --mode seqs --train True \
--epoch 10 --batch 4 --d_in 49 \
--d_hidden 512 --dim 256 --d_embedding 32 \
--n_heads 8 -h_dim 64 --integer_sequence True \
--model_save_path './models'
주요 차이점:
- 입력 데이터: PSSMs 방식은 단백질 서열 외에도 PSSMs, 2차 구조, 정보 함량을 추가로 사용합니다.
- 모델 구조: PSSMs 방식은 hidden state dimension을 늘리고, 추가 입력 데이터를 사용합니다. Protein Sequence 방식은 Transformer 모델을 사용하며, 각도의 순환적 특성을 처리하는 방식이 다릅니다.
- 목표 출력: Protein Sequence 방식은 각도의 sin과 cos 값을 예측합니다.
차이점:
두 방식은 단백질 구조 예측 문제를 해결하기 위해 서로 다른 입력 데이터와 모델 구조를 사용합니다. PSSMs 방식은 다양한 입력 데이터를 사용하여 복잡한 모델을 학습하는 반면, Protein Sequence 방식은 단백질 서열만을 사용하여 Transformer 모델을 학습합니다.
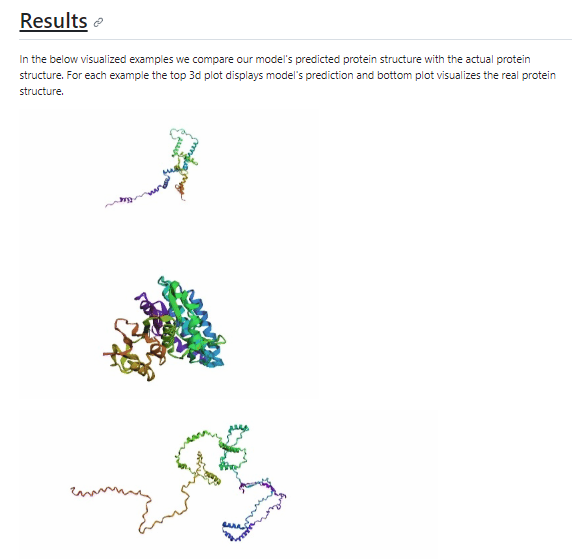
그래서 최종적으로 이 방식을 사용하니 결과가 나쁘지 않다. 라는 것인데
https://www.kaggle.com/code/basu369victor/attention-based-protein-structure-prediction
다음 과정을 실행해볼 수 있는 kaggle 에서 가져온 코드를 그대로 실행해본 결과
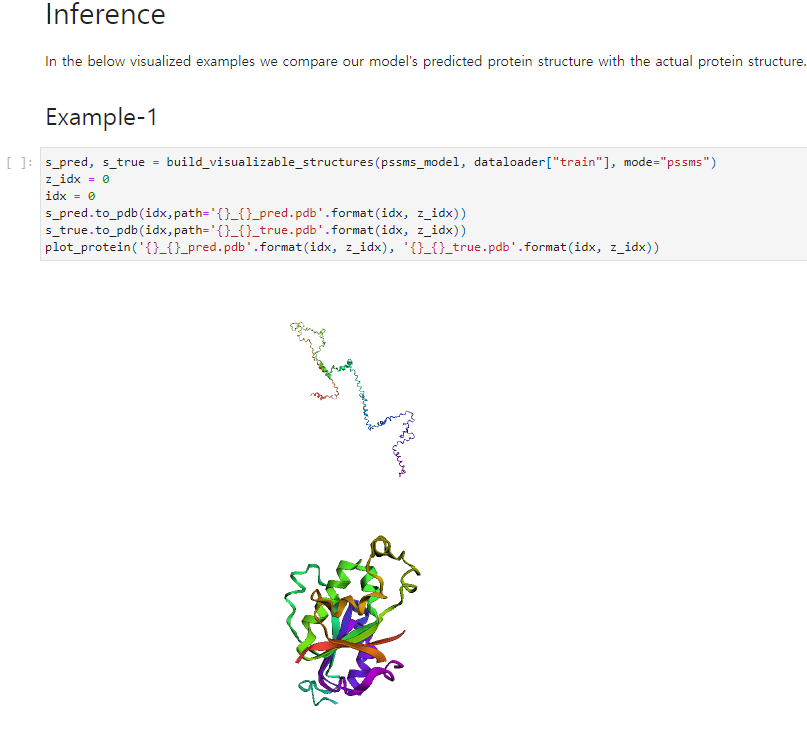
너무나도 철저하게 망해버렸습니다.
조금 더 코드를 여러 방면으로 분석하고 accuracy를 높일 방법을 찾아봐야할 것 같습니다.
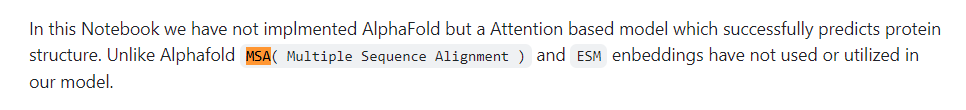
추가적으로 MSA와 ESM 이라는 단어가 나오는데 뜻은 다음과 같습니다.
MSA (Multiple Sequence Alignment)
- 정의: MSA는 여러 단백질 또는 핵산 서열을 정렬하여 서열 간의 동질성을 찾는 방법입니다. 이를 통해 진화적 관계, 공통 도메인, 보존된 아미노산 등을 파악할 수 있습니다.
- 용도:
- 단백질 구조 예측: 서열의 동질성을 기반으로 단백질의 3차원 구조를 예측합니다.
- 진화적 관계 분석: 서열 간의 유사성을 통해 진화적 관계를 분석합니다.
- 기능적 도메인 탐색: 공통된 도메인이나 모티프를 찾아 단백질의 기능을 예측합니다.
ESM (Evolutionary Scale Modeling)
- 정의: ESM은 Transformer 기반의 단백질 언어 모델로, 단백질 서열에서 구조를 예측합니다 (GitHub 페이지).
- 특징:
- ESM-2와 ESMFold는 단백질 구조 예측 작업에서 뛰어난 성능을 보입니다.
- ESMFold는 ESM-2 언어 모델을 활용하여 단백질 서열에서 직접 정확한 구조를 예측합니다.
- ESM은 단백질 설계와 관련된 여러 연구에서 활용되며, 예를 들어, "Language models generalize beyond natural proteins"와 "A high-level programming language for generative protein design" 등의 논문에서 활용되었습니다.
- 용도:
- 단백질 구조 예측: 단백질 서열에서 3차원 구조를 예측합니다.
- 단백질 설계: 새로운 단백질을 설계하는 데 활용됩니다.
- 단백질 변이 효과 예측: 단백질 변이의 효과를 예측하는 데 사용됩니다.
이 두 기술은 서로 다른 목적으로 사용되며, 단백질 서열과 구조에 대한 깊은 이해를 바탕으로 진화적 정보를 활용합니다. ESM은 Transformer 기반 모델로, 단백질의 서열과 구조를 예측하고 분석하는 데 사용되며, MSA는 여러 단백질 서열을 정렬하여 진화적 관계와 구조적 특성을 파악하는 데 활용됩니다.
참조
PPT
https://github.com/victor369basu/ProteinStructurePrediction
https://github.com/google-deepmind/alphafold#installation-and-running-your-first-prediction
https://github.com/jonathanking/sidechainnet
https://www.kaggle.com/code/basu369victor/attention-based-protein-structure-prediction
https://www.nature.com/articles/s41586-021-03819-2
https://www.researchgate.net/figure/The-basic-structure-of-the-attention-mechanism_fig2_340821036
https://tutorials.pytorch.kr/beginner/transformer_tutorial.html
https://wikidocs.net/22893
https://wikidocs.net/31379
https://velog.io/@cha-suyeon/%EB%A9%80%ED%8B%B0-%ED%97%A4%EB%93%9C-%EC%96%B4%ED%85%90%EC%85%98Multi-head-Attention-%EA%B5%AC%ED%98%84%ED%95%98%EA%B8%B0
https://wikidocs.net/162096
