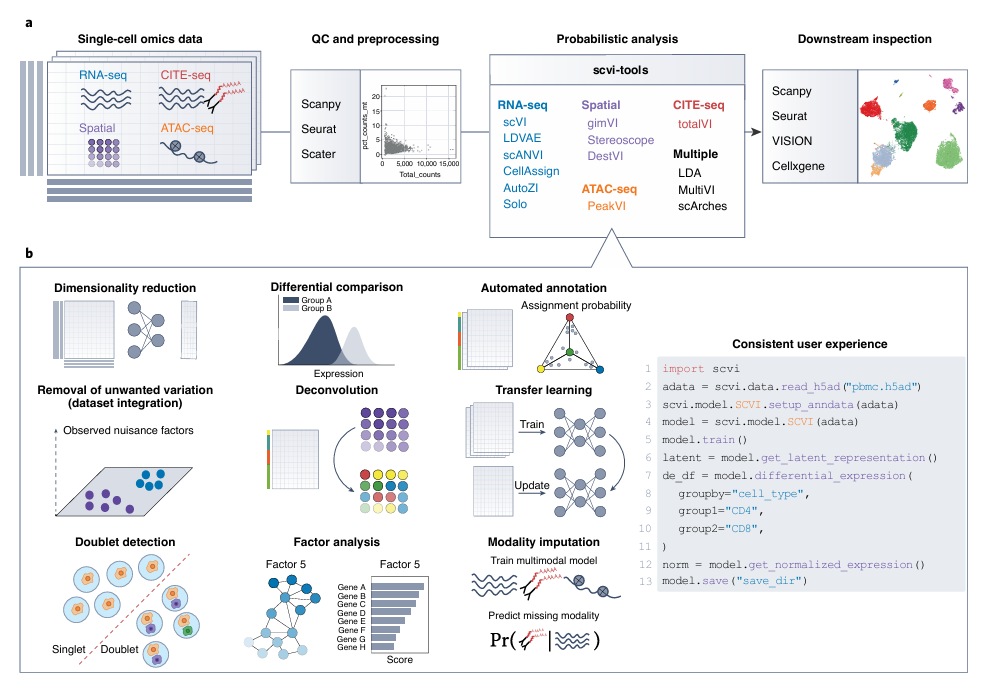
💡 scvi-tools
단일세포 오믹스 데이터를 확률적 방법으로 분석하기 위한 Python 기반 오픈소스 라이브러리인 scvi-tools
🖐 단일세포 오믹스 란?
개별 세포 하나하나의 유전자, 단백질, 염색질 등의 생물학적 정보를 분석하는 기술
큰 도시(몸 전체)가 있고, 그 안에 수많은 사람(세포)이 있어요.
기존 기술은 도시 전체의 평균 키, 몸무게, 건강상태만 알려줬다면,
단일세포 오믹스는 개개인마다 키, 몸무게, 혈압, 성격까지 각각 다르게 조사하는 기술
✅ 배경 및 필요성
1️⃣ 단일세포 오믹스 분석의 핵심 작업들
-
차원 축소: 수천 개 유전자 정보를 2~3개의 축으로 줄여 시각화나 분석을 쉽게 만듦
-
세포 클러스터링: 비슷한 유전자 표현 패턴을 가진 세포들을 자동으로 묶는 작업
-
세포 상태 주석(annotation): 각 세포가 어떤 종류인지 자동으로 이름 붙이는 과정
-
잡음/배치 효과 제거: 실험 조건이 다른 샘플 간의 불필요한 차이(배치 효과)를 제거하는 과정
-
차등 발현 분석: 두 집단 간에 특정 유전자가 얼마나 다르게 발현되는지 분석하는 과정
-
공간적 유전자 발현 패턴 탐지: 조직 내에서 어디에 어떤 유전자가 발현되는지 공간적으로 분석하는 기술
-
멀티모달 오믹스 데이터 통합 분석: RNA, 단백질, 염색질 등 다양한 생체정보를 동시에 통합 분석하는 기술
➡ 대부분 확률 기반 모델(likelihood-based models)을 활용하며,
이는 생물학적 시스템의 불확실성을 모델링하고 다양한 변이를 해석하는 데 유리함.
2️⃣ scvi-tools 소개
scvi-tools는 단일세포 오믹스 데이터의 확률론적 분석을 위한 Python 라이브러리
✅ 사용자에게
-
scRNA-seq, CITE-seq, ATAC-seq, Spatial transcriptomics 등 다양한 오믹스 데이터를 위한 통일된 인터페이스 제공
-
Scanpy, Seurat 등과의 호환성 유지
🖐 scRNA-seq (Single-cell RNA sequencing)
개별 세포마다 어떤 유전자가 얼마나 발현되는지(RNA) 측정하는 기술
🖐 CITE-seq (Cellular Indexing of Transcriptomes and Epitopes by sequencing)
RNA + 세포 표면 단백질을 동시에 측정하는 기술
🖐 ATAC-seq (Assay for Transposase-Accessible Chromatin using sequencing)
세포 DNA 중 열려 있는(chromatin accessible) 부분을 찾아, 유전자 조절 정보를 파악
🖐 Spatial transcriptomics
유전자 발현 데이터를 조직 내 위치 정보와 함께 분석하는 기술
🖐 Scanpy
Python 기반 단일세포 RNA-seq 데이터 분석 도구
🖐 Seurat
R 기반 단일세포 RNA-seq 데이터 분석 도구
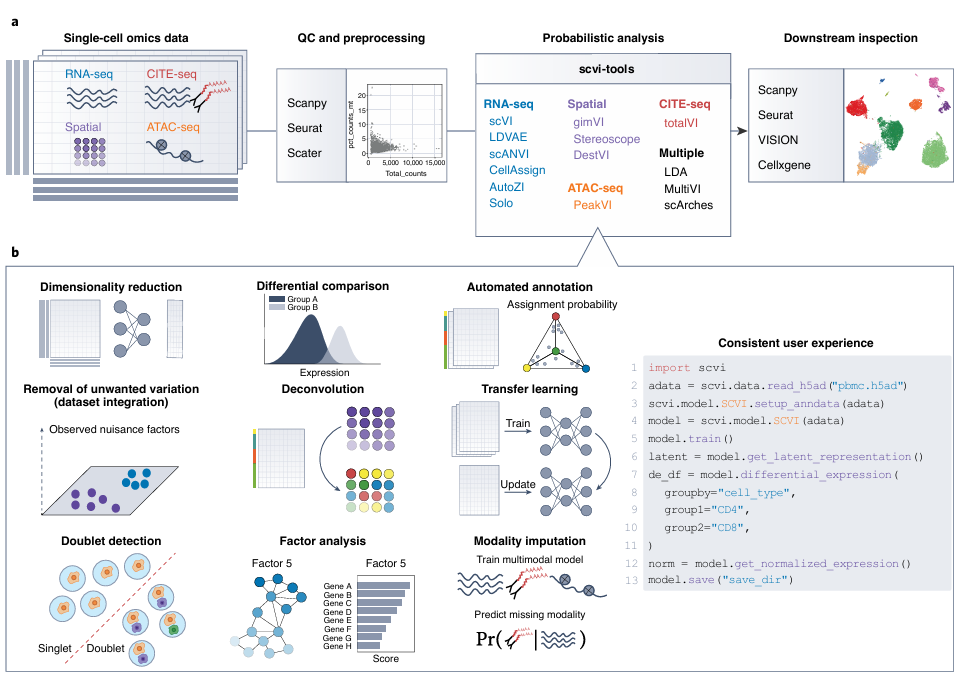
주요 기능들
-
차원 축소: 수천 개 유전자 정보를 2~3개 주요 특징만 남겨서 시각화나 분석을 쉽게 하는 과정
-
차등 발현 분석: 두 세포 그룹 간에 특정 유전자가 얼마나 다르게 발현되는지 비교
-
잡음 제거, 데이터 통합: 실험 조건에 따른 불필요한 변이(배치 효과)를 제거하고, 서로 다른 샘플을 하나로 합치는 작업
-
이중세포(doublet) 탐지: 실험 중 두 개의 세포가 하나로 측정된 이중세포(오염된 데이터)를 자동으로 식별
-
다중 모달리티 통합: RNA + 단백질 같이 서로 다른 데이터 유형을 한꺼번에 분석하는 방법
-
자동 주석: 유전자 발현 패턴을 보고 각 세포가 어떤 타입인지 자동으로 이름 붙이는 과정
-
transfer learning: 기존에 학습된 모델을 기반으로 새로운 데이터에 빠르게 적용하는 기술
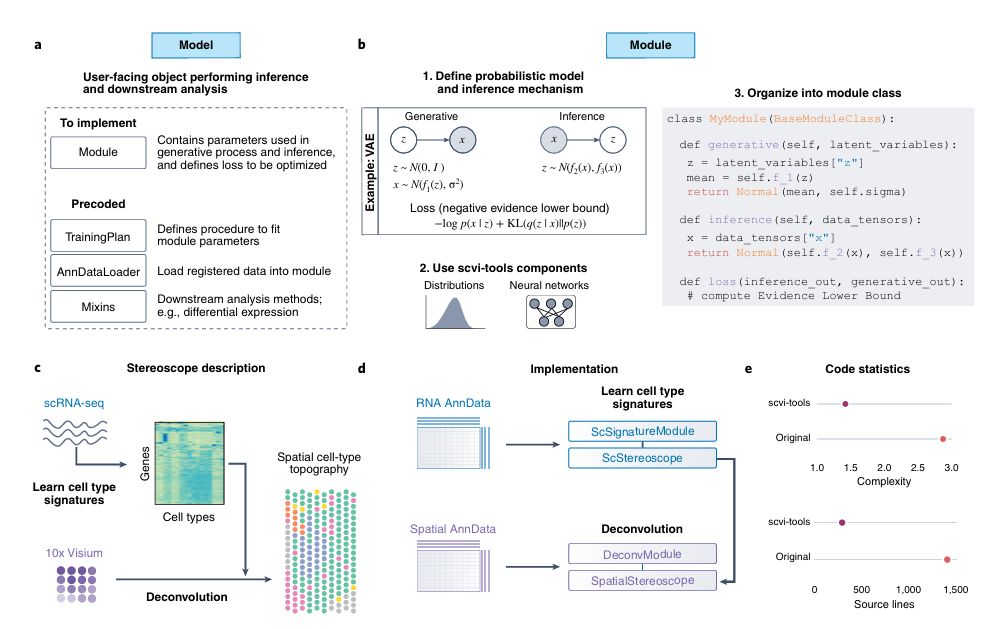
✅ 개발자에게
-
모델 구현 시 반복되는 저수준 코드들을 모듈화하여 추상화
-
AnnData, PyTorch, PyTorch Lightning, Pyro 등과 통합
-
VAE 기반 모델을 몇 줄 코드로 구현 가능
🔬 3. 기능적 확장: 실사용 예시
① 잡음 요인 제거 (Unwanted variation)
연속형 및 이산형 요인 모두 처리 가능
예: Drosophila wing development 데이터셋에서 cell cycle, sex, replicate 요인 제거
② 사전학습(reference) 모델에 새로운 샘플 추가
scArches 프레임워크 사용
전체 데이터 재학습 없이 새로운 query 데이터만 통합
예: COVID-19 환자 데이터를 totalVI 모델로 기존 면역 세포 아틀라스에 투사
📈 4. 결론 및 기여
🎯 목적
- 머신러닝 프레임워크와 생명정보 소프트웨어 생태계 간의 간극 해소
- 확률론적 모델의 표준 구현 및 배포 방식 제공
- 단일세포 분석 생태계의 발전 가속화