✅ Abstract
나노바디는 작고 안정적이지만, 나노바디 전용 설계 도구가 부족
Rosetta:
강력하지만 False Negative 가 높아 대규모 실험 스크리닝이 필요해 비용·시간·노동이 증가
NanoBinder:
Rosetta에서 뽑은 에너지 점수를 Feature 로 쓰고, 랜덤 포레스트와 SHAP 해석을 결합하여 나노바디–항원 결합 여부를 예측·설명하는 도구
49개의 다양한 나노바디로 실험 검증
-> 비결합 예측은 정확, 결합 예측도 합리적 수준
✅ Introduction
최근 단백질 설계 AI 도구(ProteinMPNN, RFdiffusion, RifDock, Hallucination, AlphaFold3, RosettaFold All-Atom 등)가 발전했지만,
(1) 항체/나노바디는 CDR 루프의 큰 가변성 때문에 특히 어려움
(2) 일반 PPI 중심의 모델은 루프 지배 상호작용을 잘 포착하지 못함
(3) 또한 항체/나노바디 구조는 PDB에서 비중이 작아 학습 편향이 생김
(4) 전용 도구(RosettaAntibody, RosettaAntibodyDesign)는 존재하나 전문가의 수작업 판단과 대규모 후보 스크리닝이 여전히 필요
(5) 최신 연구도 수천~수만 개 후보를 스크리닝해야 단 하나의 히트가 나올 수 있을 정도로 거짓 음성률이 높음
(6) 전통적 고처리량 스크리닝(효모/파지 디스플레이, FACS)은 고가의 라이브러리·장비와 숙련 인력을 요구하고, 그럼에도 유망 후보를 놓칠 위험이 있어 효율적 계산 필터가 필수
(7) 기존 ML 시도(NbX, 딥러닝 기반 포즈 랭킹 등)는 일반 항체·일반 PPI에 최적화되어 나노바디에는 덜 효과적
제안: Rosetta 점수(구조 에너지)를 피처로 실험적으로 검증된 결합/비결합 복합체에서 ML을 학습
→ 나노바디 설계 단계의 결합 확률을 예측해 사람 손의 선별 부담과 실험 규모를 축소
모델·도구 개요
분류기는 랜덤 포레스트 채택:
비선형 관계와 피처 상호작용을 잘 다뤄 나노바디–항원 특징/결합 확률의 복잡한 연관성에 적합
✅ Result
✅ 핵심 아이디어와 워크플로우
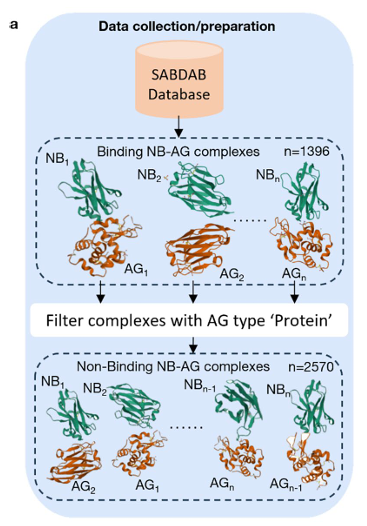
SAbDab에서 결합 complex 1,396개를 모으고, 정렬 기반 전략으로 비결합 복합체 2,570개를 합성해 학습용 ‘양/음성’ 데이터를 구성(총 3,966개)
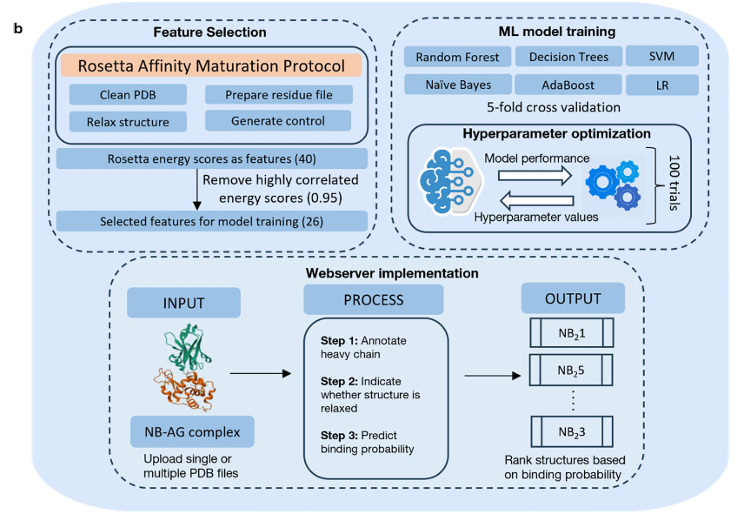
Rosetta(주로 RosettaAntibody/Design)로부터 나온 에너지 점수를 구조적 특징으로 사용
상관계수 0.95 초과 항목으로 필터링 해 최종 26개 특징을 선별
(예: dG_cross, dG_cross/dSASA×100, per_residue_energy_int 등).
학습은 RF, SVM, DT, AdaBoost, LR, Naive Bayes 등 다양한 분류기를 동일한 계층화 5-fold 교차검증과 랜덤 탐색 하이퍼파라미터 최적화로 비교
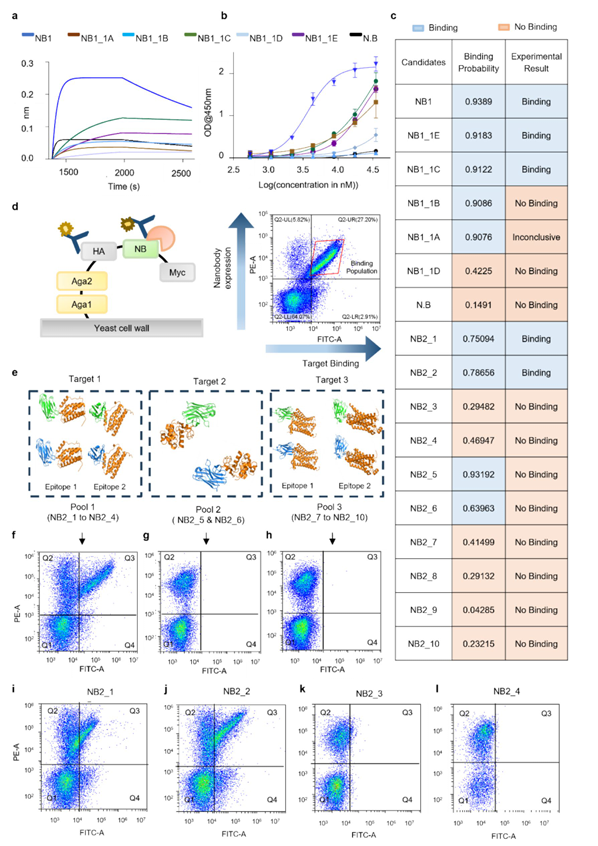
실험 검증은 두 단계 설계:
(1) 기존 항체에서 CDR grafting으로 유래한 나노바디를 Rosetta로 정제·설계 후 후보 5개 선별
(2) 다섯 개 epitope across 세 항원 등 다양성을 높인 세트로 확장
결합 평가는 Yeast Surface Display + FACS로 수행
✅ 교차검증 성능
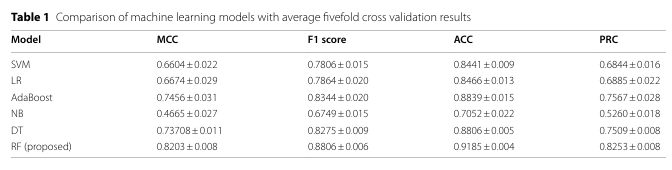
Random Forest(RF) 가 모든 모델 중 최고:
MCC 0.8203, F1 0.8806, ACC 0.9185, PRC 0.8253.
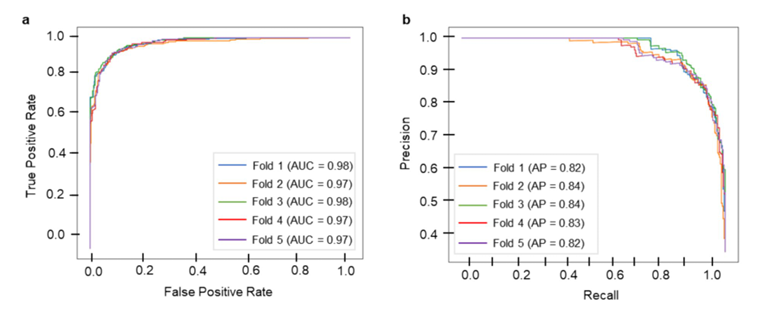
PR-curve와 ROC-curve는 5개 fold 모두에서 양호
✅ SHAP 해석: 어떤 특성이 결합/비결합을 가르는가
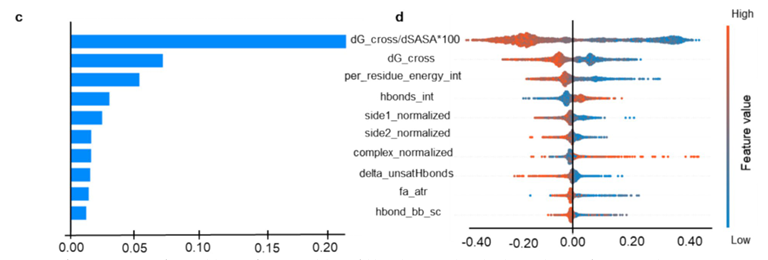
가장 영향력이 큰 특성: dG_cross/dSASA×100 (자유에너지 변화 dG를 계면 dSASA로 나눈 뒤 100배 스케일)
값이 높을수록 비결합 쪽(양의 SHAP), 낮을수록 결합 쪽(음의 SHAP)으로 작용
-> 생물학적 기대(더 음의 dG가 결합 유리)와 일치
두 번째 핵심 특성: dG_cross(계면 자유에너지)
더 음수(강한 결합)일수록 결합 예측을 끌어올림
그 밖의 유효 특성:
per_residue_energy_int, hbonds_int(잔기 수준 에너지·수소결합)
side1_normalized, side2_normalized(각 면 평균 잔기당 에너지)
complex_normalized(복합체 평균 잔기당 에너지)
delta_unsatHbonds(매몰된 미충족 H-bond 수), fa_atr(원자 간 인력)
hbond_bb_sc(backbone–sidechain H-bond)
✅ 실험적 검증
Set 1, 2
Set 3
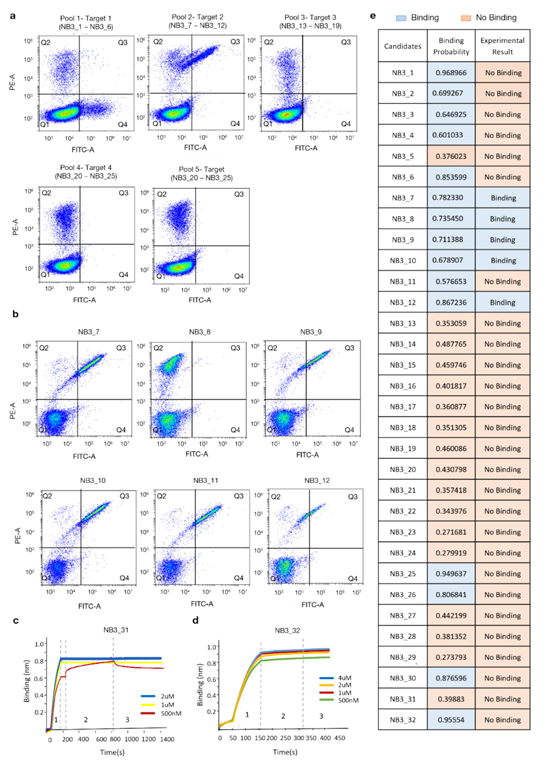
✅ 예측–실험 상관 코멘트
예측 확률 ≥0.5 → 잠재적 결합자, <0.5 → 비결합자로 정의
Set1 약 71% 상관(0.9↑ 예측 6개 중 3개 결합, 최저 확률 후보는 비결합)
Set2 약 80% 상관(0.5↑ 4개 중 2개 결합, 0.5↓ 6개 전부 비결합)
Set3 약 68% 상관(0.5↑ 15개 중 5개 결합, 17개 비결합 적중)
세트 합산: 비결합 100% 정확, 결합 적중률 43%
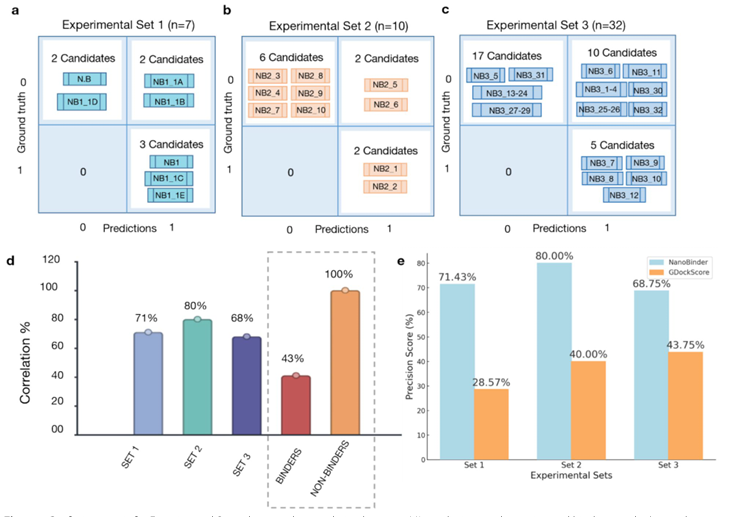
GDockScore와의 비교
동일 임계(0.5)에서, NanoBinder가 모든 세트에서 GDockScore보다 우수
왜 NanoBinder가 더 잘하나?
GDockScore는 나노바디-항원(NB-AG) 복합체로 학습된 점수가 아니며, NB-AG는 다른 PPI와 결합부위의 진화적 보존성 등에서 생물학적으로 구별됨
그래서 나노바디 특유 결합 패턴을 충분히 포착하지 못함
✅ Discussion
Rosetta 에너지 점수를 특징으로 한 ML 프레임워크를 제안했고, 랜덤 포레스트(RF) 가 결합/비결합을 잘 가려 MCC 0.8203을 달성
Rosetta의 구조적 통찰과 ML 결합의 유효성을 보였으며, 특징 선택에서 dG_cross/dSASA×100이 가장 큰 기여
✅ 한계와 오류 원인
세트 1처럼 서열·구조가 거의 같은 나노바디들 사이의 아주 작은 구조 차이에는 민감도가 떨어짐
→ 미세한 구조 차 구분에 약함
거짓 양성 사례 존재: 예측 확률이 매우 높아도(~0.9) 실험에선 결합하지 않은 경우가 있었음
→
(1) 비결합체가 진짜 결합체와 유사한 Rosetta 에너지 프로파일을 보여 모델이 헷갈렸고(dG_cross/dSASA×100, hbonds_int, per_residue_energy_int가 유사)
(2) 일부 비결합 구조 모티프가 데이터에 충분히 대표되지 않아 모델이 그 후보들을 정확히 거르지 못했으며
(3) 도킹 단계에서는 결합처럼 보이지만 실험적으로 안정 결합으로 이어지지 않는 상호작용이 있기 때문이라고 분석
프레임워크 영역의 돌연변이가 CDR 루프 배치를 미세하게 바꾸는데, Rosetta의 루프 예측 한계로 이런 변화가 구조·점수에 충분히 반영되지 않음
→ 실제 결합력 상실이 있어도 모델이 높은 결합 확률을 줄 수 있음
왜 ‘비결합’ 예측이 특히 강한가
실제 단백질 affinity maturation 환경에선 대량 후보 중 비결합자가 다수이므로, 이를 반영해 음성(비결합) 비율을 더 높게 데이터셋을 구성
덕분에 모델은 비결합 식별에 특화되었고, 향후 예측에서도 비결합을 잘 걸러낼 수 있음
(실험 스크리닝 비용·시간 절감에 유리)
✅ Method
1) Dataset collection & preparation
출처: SAbDab에서 실험적으로 결합이 확인된 나노바디–항원(NB-AG) 복합체 1,396개를 수집
PDB ID, 나노바디 heavy chain, 항원 체인, 항원 타입을 추출
항원 타입이 Protein인 경우만 사용(탄수화물·펩타이드·핵산·헵텐 제외)
비결합(non-binding) 복합체 구성:
결합 복합체 2개를 무작위 선택해 PyMOL로 각각 로드
→ annotate_v 스크립트로 heavy chain 선택 후 align으로 heavy-chain 정합
정합 RMSD < 2 Å일 때, 한 복합체의 나노바디와 다른 복합체의 항원을 합쳐 새 NB-AG로 내보냄
각 쌍에서 2개의 비결합을 생성. 중복 방지를 위해 한 결합 복합체는 한 번만 사용
모든 조합을 반복해 비결합 2,570개를 만듦
최종 데이터셋은 결합 1,396 + 비결합 2,570 = 총 3,966개 구조.
2) Data preprocessing & Rosetta protocol
각 PDB에서 물분자·불필요 리간드 제거
나노바디 구조의 constant domain(CH) 제거. 체인명은 H(나노바디), A(항원)으로 통일
Rosetta에 NATAA(원래 측쇄 유지, repack)로 계면 잔기 전부 리팩하도록 residue 파일 준비(새 나노바디 설계가 목적이 아니라 에너지 스코어 추출이 목적)
Relax로 백본 φ–ψ와 측쇄 충돌 최소화(구조 1개당 단 한 번의 relax)
이후 아미노산 설계는 수행하지 않음(네이티브 서열의 에너지에 가깝게 유지)
결과로 에너지 스코어 40종을 생성해 ML 특징으로 사용
3) Feature selection
Rosetta 40개 스코어 중 packstat, sc_value는 전 샘플 0이라 제거, total_score는 다른 항목의 가중 합이라 다중공선성 때문에 제외
→ 37개로 축소
상관 제거: 모든 특징 쌍의 피어슨 상관(|r|)을 계산, 임계 0.95 초과 시 중복으로 간주해 제거(양/음 상관 모두 동일 취급)
이렇게 중복 변수를 제거해 일반화 성능을 높이도록 설계
최종적으로 26개 특징을 사용:
complex_normalized, dG_cross, dG_cross/dSASAx100, dSASA_hphobic, dSASA_int, dSASA_polar, delta_unsatHbonds, dslf_fa13, fa_atr, hbond_E_fraction, hbond_bb_sc, hbond_lr_bb, hbond_sc, hbond_sr_bb, hbonds_int, nres_int, omega, per_residue_energy_int, pro_close, rama_prepro, ref, side1_normalized, side1_score, side2_normalized, side2_score, yhh_planarity.
4) Model training
계층화 5-fold 교차검증(StratifiedKFold): 고정 random state와 셔플 사용
imblearn 파이프라인으로 각 fold에서 SMOTETomek(SMOTE + Tomek links) 훈련 세트에만 적용하여 클래스 불균형 완화 후 학습