✅ Abstract
단백질의 단일 점돌연변이가 안정성(ΔΔG)에 미치는 영향을 더 빠르고 정확하게 예측하기 위해, 서열 언어모델 임베딩과 3D 그래프 기반 모델을 결합한 새 방법 ProSTAGE를 제안
보편적으로 쓰이던 학습셋(S2648)의 거의 두 배 규모 데이터로 훈련했고, 여러 독립 벤치마크에서 기존 SOTA들을 안정적으로 앞섬
✅ Introduction
단백질 열역학적 안정성은 구조·기능·발현·용해도에 직결되고, 돌연변이는 이를 떨어뜨려 접힘 실패·질환을 유발할 수 있음
그래서 변이가 안정성(ΔΔG)에 미치는 영향을 예측하는 계산 방법은 단백질 설계·정밀의학에 필수
AlphaFold의 등장은 “실험 구조가 없어도 구조 기반 접근”을 가능하게 했고, 동시에 NLP가 서열의 문맥 의존성을 잘 잡는다는 점을 보여줌
문제는 데이터 크기
표준 훈련 세트(S2648, Q3421)는 “몇 천 개 수준”이라 딥러닝을 충분히 돌리기엔 작아 많은 방법들이 여전히 얕은 ML에 머뭄
이 논문이 제안하는 해법
ProSTAGE:
구조와 서열 임베딩을 융합하는 딥러닝 접근
그래프 기반 기법과 언어모델을 결합해 단일 점돌연변이의 안정성 변화를 예측
언어모델 임베딩을 쓰면 별도의 도메인 지식으로 피처를 설계할 필요가 없음
그래프 쪽은 변이 주변의 잔기 상호작용(공간적 근접) 을 담는 spatial node feature를 제안하고, 노드 입력으로 사전학습 단백질 임베딩을 넣어 GCN이 처리하게 함
훈련 데이터는 여러 소스에서 모아 S2648의 약 2배로 확대했고, 여러 독립 벤치마크에서 기존 SOTA를 일관되게 상회
✅ Materials & Methods
1) Data set 수집·정제 (Training / Test)
훈련 셋(S11304)
- 출처: MPTherm, ProThermDB, ThermoMutDB, FireProtDB 등 4개 최신 DB에서 실험 ΔΔG가 있는 단일 점돌연변이만 모음
- 중복 제거(non-redundant) 규칙을 적용
- 반대칭(antisymmetry) 보장: 변이 A→B의 ΔΔG와 B→A의 ΔΔG가 부호가 반대가 되도록 쌍을 구성(ΔΔG(A/B)=−ΔΔG(B/A))
- 규모: 11,304 변이 / 318단백질(기존 표준 S2648의 약 2배 수준)
- 안정화/불안정화의 클래스 불균형을 완화하려고 분포를 조정
블라인드 테스트 셋
① S669
- 가장 널리 쓰이는 엄격 블라인드 세트
- ΔΔG가 있는 669개 단일 변이로 구성(훈련셋과 단백질·변이 중복 없음)
② Tm262 / Tm108
- 이 두 세트는 ΔTm(용융온도 변화)만 제공되는 변이로 구성(같은 출처 기준 규칙으로 선별)
- |ΔTm| ≥ 2 °C 같은 컷오프를 적용하여, 안정화/불안정화 이진 분류 성능을 공정하게 비교할 수 있게 함
③ DMS(PTEN, TPMT)
- CAGI5에서 공개된 깊은 변이주사(DMS) 점수(PTEN/TPMT 각 수천 변이)
- 여기서는 연속 점수 상관(Pearson)으로 평가(ΔΔG와 DMS fitness의 일치도)
2) 그래프 신경망(GCN) 예측기 설계
그래프 정의(G = (V,E))
노드 V: 변이 잔기 주변 10 Å 이내의 잔기들(변이 위치 포함)
엣지 E: 두 잔기의 Cα 간 거리가 ≤10 Å이면 연결(공간 인접 기반 Adjacency Matrix A)
노드 특징 X (핵심 포인트): 전통적 one-hot이나 선형 이웃(좌/우 k-mer)이 아니라, PLM 임베딩 기반으로 구성
ProtT5-XL-UniRef50의 사전학습 임베딩을 고정(frozen)해 사용
각 노드의 특징은 WT 서열 임베딩 ⊕ Mutant 서열 임베딩을 concat한 벡터
차원: F = 2048 (= 1024×2)
노드 수 N은 반경 10 Å 내 잔기 수
-> 실제 변이는 주변 상호작용을 바꾸므로, 변이 전후 문맥을 함께 보여주는 게 유리
GCN 본체
GraphConv(3층, 각 64 유닛) → 각 층 출력을 concat → 풀링(global/readout)
여기에 지식 기반 부가 특징(AKB)을 더해 MLP(FC 3층)로 ΔΔG 회귀 출력
AKB(추가 지식 특징)
구조/진화 기반의 상대 용매접근도(RSA), 보존도, 이차구조, 접촉수/지오메트리 등
목적: PLM+GCN이 담지 못한 설명적·물리적 단서를 보강
3) 이 설계가 갖는 의도/의미
왜 공간 그래프?
변이는 국소 상호작용망을 바꾸므로, 단순 k-mer보다 3D 이웃이 합리적
왜 WT⊕Mut 임베딩?
변이의 전후 차이를 모델이 직접 학습하도록 설계(단일 임베딩보다 변화 감지에 유리)
왜 AKB 추가?
PLM/GCN이 놓칠 수 있는 물리·보존 신호를 보강해 일반화 개선
✅ Result
모델 개요 (Figure 1)
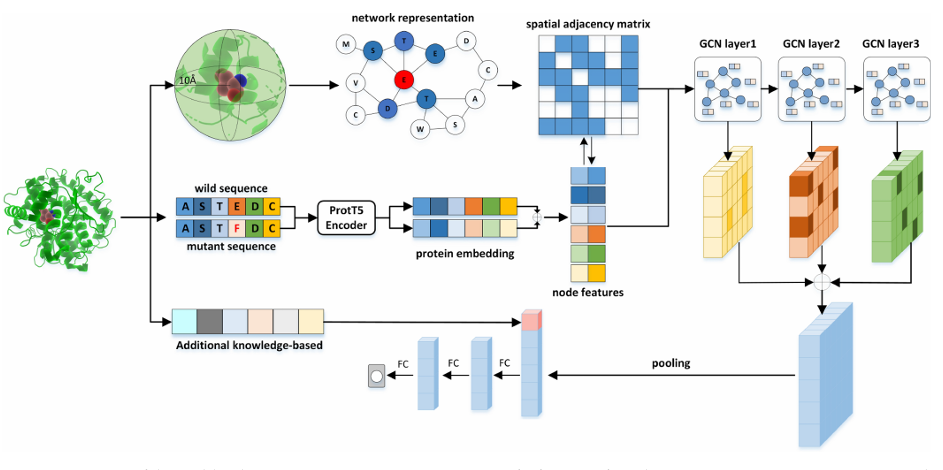
입력:
변이 단백질의 야생형(wild)·변이형(mutant) 서열을 ProtT5-XL-UniRef50로 임베딩(각 1024차원)
→ 두 임베딩을 concat해 2048차원 노드 특징으로 사용
공간 그래프:
변이 지점에서 10 Å 이내 잔기들을 노드로 잡고, 거리 기반 spatial adjacency로 엣지를 만듦
GCN 본체:
GraphConv 3층(각 층은 이전 층 임베딩과 공간 인접행렬을 함께 사용) → 세 층 임베딩을 concat → pooling
여기에 추가 지식 기반 특징(AKB)을 더해 FC 3층을 거쳐 ΔΔG 회귀를 출력
교차검증 성능 (Figure 2)
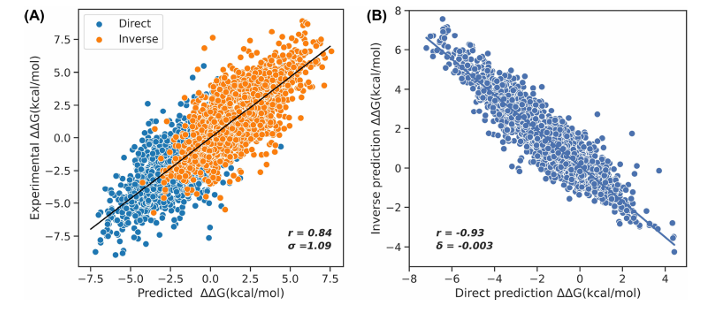
5-fold CV(검증 파트 20%)에서 PCC = 0.84, RMSE = 1.09 kcal/mol.
“5-fold CV(검증 파트 20%)”
훈련셋 S11304을 5등분해서, 매번 1등분(=20%)을 검증용, 나머지 4등분(=80%)을 학습용으로 써서 총 5번 학습→예측→평균 성능을 냄
반대칭(antisymmetry) 점검:
직접 변이 vs 역변이 예측의 상관이 r = −0.93, bias(δ) ≈ −0.003 kcal/mol로 “거의 완벽히” 반대 부호를 만족. (그림 2B)
LOPOCV(단백질 단위 제외 검증)에서도 PCC = 0.75, RMSE = 1.34 kcal/mol로 일반화 성능을 확인.
비교 실험 세팅
비교 대상:
PROST, PremPS, ACDC-NN /
-SEQ, DDGun /
3D, ThermoNet, Dynamut, Rosetta (Pancotti et al.에서 상위권 모델들)로 한정
평가 셋은 S669, Tm262, PTEN, TPMT의 4개 블라인드 셋을 사용
S669 공정 비교 방식:
(1) 학습셋에서 S669와 상동인 변이를 제거,
(2) 혹은 S2648만으로 학습한 버전을 함께 보고
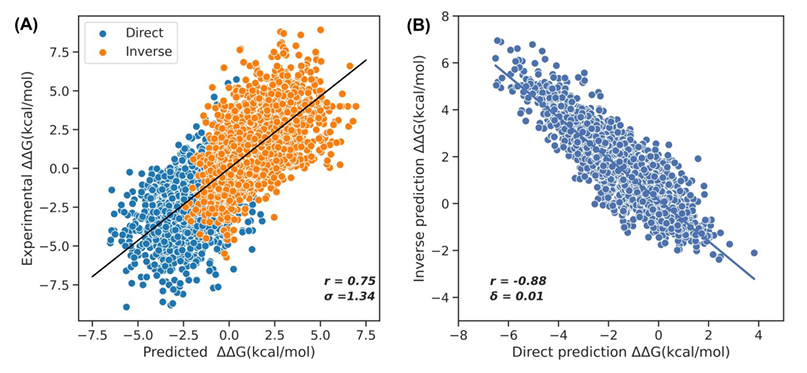
S669(ΔΔG 회귀) 결과 — Table 1
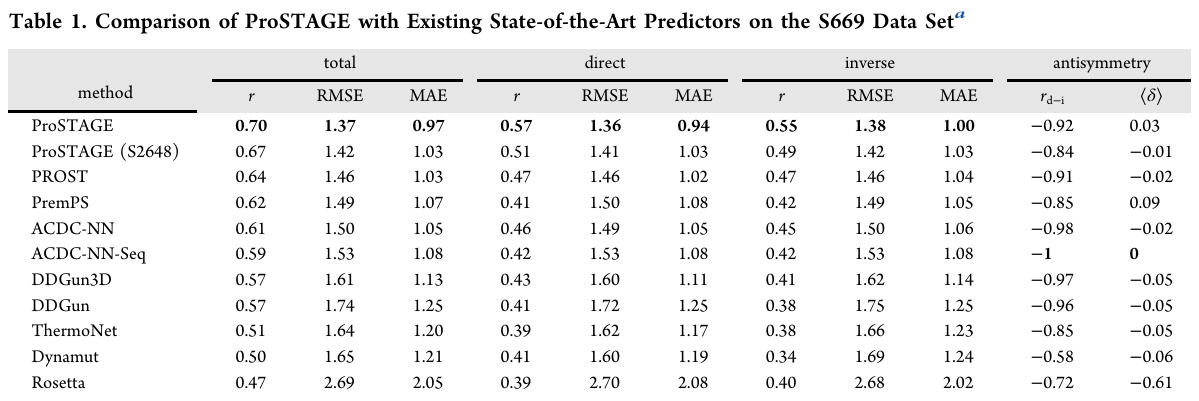
총합 성능:
ProSTAGE r=0.70, RMSE=1.37, MAE=0.97로 1위
(2위 PROST r=0.64, RMSE=1.46, MAE=1.03)
직접 변이: r=0.57, RMSE=1.36, MAE=0.94(최고).
역변이: r=0.55, RMSE=1.38, MAE=1.00(경쟁 모델 대비 뚜렷한 우위).
반대칭 검증:
예측한 직접/역변이 사이 상관 r_{d−i} = −0.92,
바이어스 ⟨δ⟩ = 0.03(이상적 반부호 관계에 매우 근접)
S2648로만 학습한 ProSTAGE도 r=0.67, RMSE=1.42, MAE=1.03으로 타 모델을 상회
→ 모델 구조+대형 데이터의 결합이 성능을 끌어올림
Tm262 / Tm108(안정/불안정 이진 분류) — Table 2
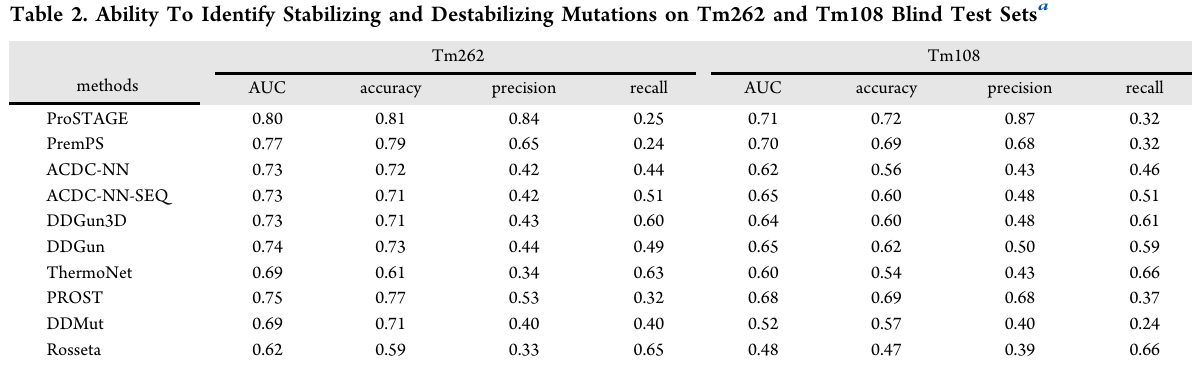
Tm262: AUC 0.80, ACC 0.81, Precision 0.84, Recall 0.25로 최고(데이터 특성상 ΔTm↔ΔΔG 비선형이라 분류로 평가)
Tm108(Tm262의 ≤25% ID 서브셋; 일반화 평가): AUC 0.71, ACC 0.72, Precision 0.87, Recall 0.32 (1위)
DMS 대회 데이터(PTEN/TPMT, 포화 변이 스코어 상관) — Table 3
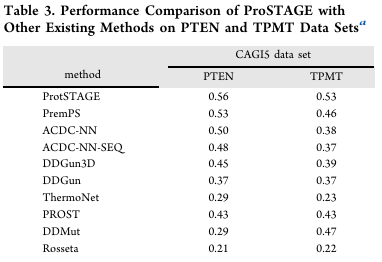
PCC(PTEN/TPMT): ProSTAGE 0.56 / 0.53으로 1위
(다른 모델 0.21–0.53 / 0.22–0.46)
AF2 예측 구조로도 잘 동작해 실험 구조 불필요성을 보여줌
소거(Ablation) — Table 4
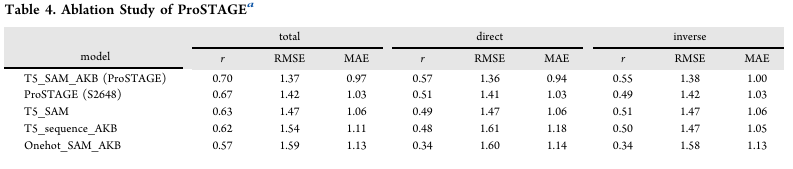
완전 모델(T5_SAM_AKB):
r=0.70/0.57/0.55(총합/직접/역), RMSE=1.37/1.36/1.38.
임베딩 핵심성: T5 임베딩 제거(Onehot_SAM_AKB) 시 총합 r=0.57로 급락.
T5_SAM에서 AKB 제거하면 r=0.63
T5_sequence_AKB(공간 그래프 → 서열 인접 그래프로 대체)도 r=0.62로 하락
⇒ T5 임베딩이 가장 큰 기여, SAM(공간 인접)·AKB는 소폭 향상
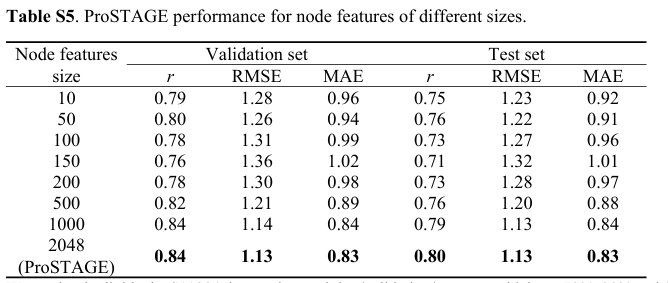
노드 차원(10–2048) 테스트: 2048에서도 과적합 위험 없음을 Table S5로 확인.
데이터 풍부함 효과:
학습셋을 S2648로 축소 시 성능 저하 → 데이터 리치가 중요
또한 진화정보 의존도 완화(고아 단백질·빠른 진화 단백질에도 유리) 주장
✅ Discussion
연구 흐름의 변화와 한계
안정성 예측 분야는 오래된 얕은 ML에서 딥러닝 중심으로 이동했지만(S2648, Q3421 같은) 표준 학습셋 규모가 작아 딥러닝 요구치를 충족하지 못해 왔음
한편 AI 기반 예측 구조(AF2 등)의 등장은 구조 기반/서열 기반 방법 간 입력 제약을 완화해, 문제에 따라 방식을 선택할 수 있게 함
저자들은 ProSTAGE를 제안
(1) 직접·역변이를 함께 학습해 반대칭(antisymmetry)을 만족
(2) 단백질 임베딩(PLM)과 공간 인접행렬(SAM)을 결합해 국소 3D 상호작용과 장거리 서열 문맥을 동시에 포착
(3) 언어모델 임베딩을 쓰므로 별도의 도메인 지식 기반 피처 설계가 불필요
어블레이션으로 임베딩이 모델 정확도 향상에 큰 기여를 함을 보임
또한 학습셋을 S2648의 2배 규모로 확장
효과 및 일반화 주장
저자들은 ProSTAGE가 여러 블라인드 셋에서 기존 방법보다 우수(정확도·AUC 모두)하며 과적합 위험을 낮춘다고 요약
특히 AlphaFold2 예측 구조를 입력으로 써도 높은 정확도를 유지해, 실험 구조 없이도 확장 가능성이 크다고 강조