✅ Introduction
✅ 1. 왜 PropEn이 필요한가?
(1) 항체 lead 최적화의 어려움
컴퓨터로 항체 후보(lead)를 설계·개선하는 과정
고품질 데이터가 적음
실험으로 정확히 측정된 항체 데이터가 많지 않음
실험 검증이 비싸고 느림
여러 개발 가능성(developability) 특성을 동시에 최적화해야 함
예: 결합 친화도(affinity), 발현량(yield), 안정성, 용해도 등
(2) 현재 실험 워크플로우의 구조적 한계
초기에 phage / ribosome / yeast display 같은 high-throughput 실험을 많이 돌림
장점: 수많은 변이체를 한 번에 스크리닝 가능 → 데이터 양은 큼
단점: 측정값이 노이즈가 크고 부정확함
이 실험들은 정확하지만, 속도가 느리고 규모 확장이 안 됨
(3) 딥러닝 · LLM 접근법의 문제
데이터가 적고 노이즈가 크며, 기능에 중요한 부분이 sequence/structure 상에서 듬성듬성 나타남
-> 많고 깔끔한 데이터를 필요로 하는 DL 모델의 성능을 크게 떨어뜨림
✅ 2. PropEn(Property Enhancer)의 핵심 아이디어
(1) PropEn 소개
데이터 효율적(data-efficient) 프레임워크
훈련 데이터가 적고(수백 개 수준),
여러 종류의 실험 데이터가 뒤섞여 있는 low-data, heterogeneous regime에서 잘 동작하도록 설계
여러 항체 특성을 동시에 최적화 가능
(2) Matching-based augmentation (매칭 기반 증강)
PropEn의 핵심 트릭은 matching-based augmentation
가지고 있는 항체 데이터 안에서 서로 매우 비슷하지만, 특정 property 값이 더 좋은 것을 짝(pair)으로 만듦
예: 두 서열이 몇 개 아미노산만 다르는데, B가 A보다 binding affinity가 더 좋다.
이렇게 해서 “sequence pairs”를 만듦
각 pair는 약간의 돌연변이만 존재하고,
pair 안에서 두 번째 서열이 항상 property가 더 좋은 쪽으로 설정
이걸 대량으로 만들면, 원래는 n개밖에 없던 데이터를
“정렬된 쌍(ordered pairs)” 수만큼 크게 확장할 수 있음
→ 수십·수백 배로 데이터가 늘어난 효과
이 구조 때문에, 각 pair는 “어떻게 바뀌면 property가 좋아지는지”에 대한 암묵적 최적화 신호를 제공
(3) PropEn 모델이 학습하는 방식
PropEn은 encoder-decoder 구조의 deep model을 사용
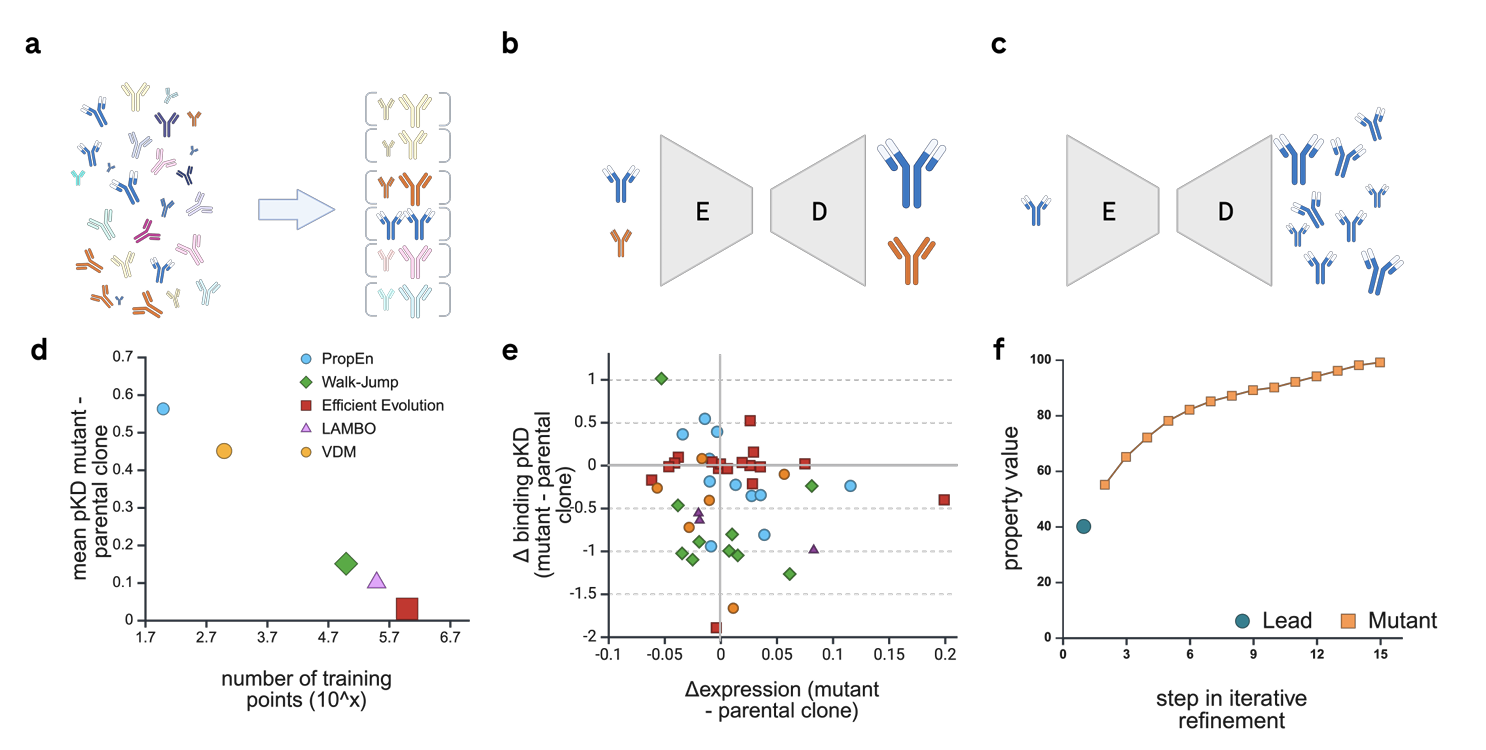
Fig. 1a – 매칭 단계
먼저, 훈련 데이터에 있는 각 항체를
“가장 비슷하지만 property가 더 좋은 항체”와 매칭
예: A(나쁨) ↔ B(더 좋음).
Fig. 1b – 학습 단계
이렇게 만든 쌍(A→B)을 이용해
“입력: 낮은 property 항체, 출력: 높은 property 항체”가 되도록 encoder-decoder를 학습
loss는 단순 reconstruction loss (입력 A를 B로 재구성하도록)
왜 이게 좋은가?
A와 B는 서열이 거의 같으므로
모델은 “항체라는 분포(유효한 후보 공간)” 안에서만 움직이도록 학습됨
동시에, 항상 “더 좋은 쪽”으로 매핑하도록 배운다 → implicit optimization.
Fig. 1c – 생성 단계
학습이 끝나면, 임의의 parental clone(시드 항체)을 넣고
수천 개의 변이체(mutants)를 샘플링해서 만들어낼 수 있음
이 변이체들은 원래 항체와 비슷하지만,
학습된 매핑 덕분에 property가 향상되도록 쏠려 있게 됨
Fig. 1f – 반복 refinement
생성한 mutant 중에서 좋은 것들을 다시 모델에 넣어
여러 라운드(iterative refinement)로 계속 개선해 나갈 수 있음
→ 한 번만 돌리는 게 아니라,
“좋아진 항체 → 더 좋아진 항체”로 트랙을 따라가는 최적화 궤적(trajectory) 생성
✅ 3. PropEn의 성능 및 기존 기법과의 비교
(1) 적은 데이터 + 작은 모델로 높은 성능
PropEn은 훈련 샘플 수가 훨씬 적어도
기존 diffusion 모델들(LAMBO, VDM)과 LLM 기반 Efficient Evolution보다 더 큰 affinity 향상을 보여줌 (Fig. 1d)
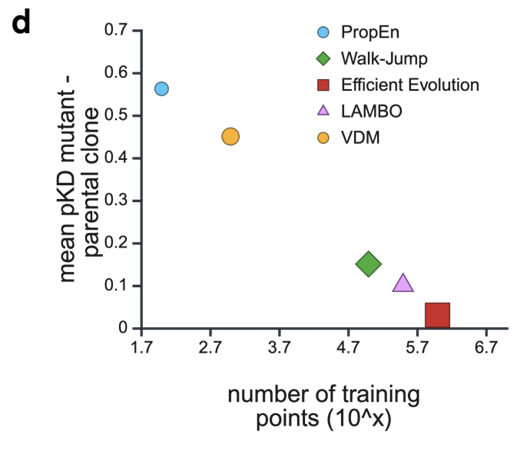
동시에, 사용하는 모델의 파라미터 수(모델 capacity)가 훨씬 작음
(2) 여러 property를 동시에 올림
실험(in vitro) 결과에서, PropEn은 네 개의 서로 다른 항원 타깃 , 아홉 개의 리드 항체에 대해,
결합 친화도(affinity)와 발현량(expression)을 동시에 개선 (Fig. 1e)
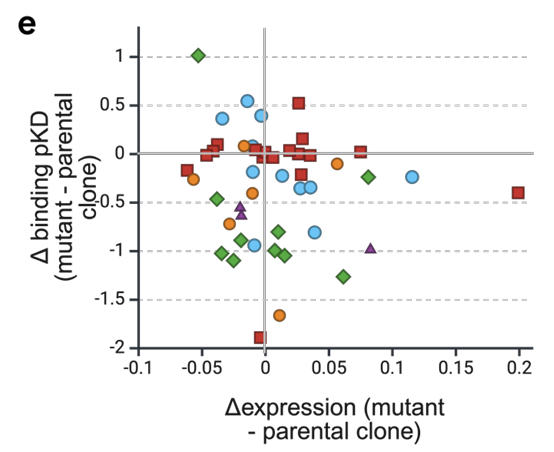
전체적으로 10–39배(10–39×)의 affinity gain을 달성했다고 abstract에 명시
(3) Pareto front와의 관계
다양한 목표(affinity, expression 등)를 동시에 최적화할 때는
서로 트레이드-오프가 생김
예: binding을 올리면 expression이 떨어질 수도 있음.
Pareto front:
더 이상 어떤 목표도 악화시키지 않고는 다른 목표를 개선할 수 없는 “효율의 최전선”.
논문은 PropEn이 affinity, expression 같은 복합 목표를 다루면서도 baseline 방법들보다 Pareto front에 더 가까운 설계들을 제안한다고 설명
✅ 4. PropEn의 세 가지 주요 장점
(1) Surrogate model이 필요 없음
다른 ML-기반 최적화에서는 보통 “property를 예측하는 별도의 모델(surrogate)”을 만들어, 그 모델을 최대화하는 쪽으로 설계하는 방식을 씀
하지만 이런 surrogate는 noisy, 적은 데이터를 가지고 학습해야 해서 부정확해지기 쉽고, 그 오류 때문에 최적화 방향이 틀어지기 쉬움
PropEn은 이런 별도 surrogate 없이, 직접 “나쁜 → 좋은” 매핑을 학습
즉, surrogate 하나를 건너뛰어서 ML-driven optimization에서의 중요한 병목을 제거했다는 의미
(2) 기능적 관련성 보장
PropEn은 항체 space 전체를 막 돌아다니지 않고, “viable(실제로 말이 되는) 분자들 근처”에서만 생성하도록 설계
학습 데이터 쌍 A–B가 서로 매우 비슷한 in-distribution 후보들이기 때문
모델은 “실제 실험에서 관측된 항체 형태” 근처에서만 이동하도록 학습됨
결과적으로, 만들어진 설계들이 생물학·화학적으로 터무니없는 구조가 아니라, 실제로 표현 가능한, 기능적인 항체 후보가 될 가능성이 높음
(3) 동시 multi-property 최적화
PropEn은 하나의 종합 점수(ranking score)를 최적화함
이 점수는 여러 property(affinity, expression 등)를 함께 반영하는 multivariate ranking score
따라서 모델이 샘플링하는 후보들은 여러 property 관점에서 Pareto front 근처에 위치하도록 유도됨
쉽게 말하면, “결합만 좋고 발현은 망한 항체”가 아니라, “여러 조건을 적당히 다 만족하는 항체”를 찾는 데 유리
✅ 5. Figure 2 – 실제 실험(in vitro) 결과
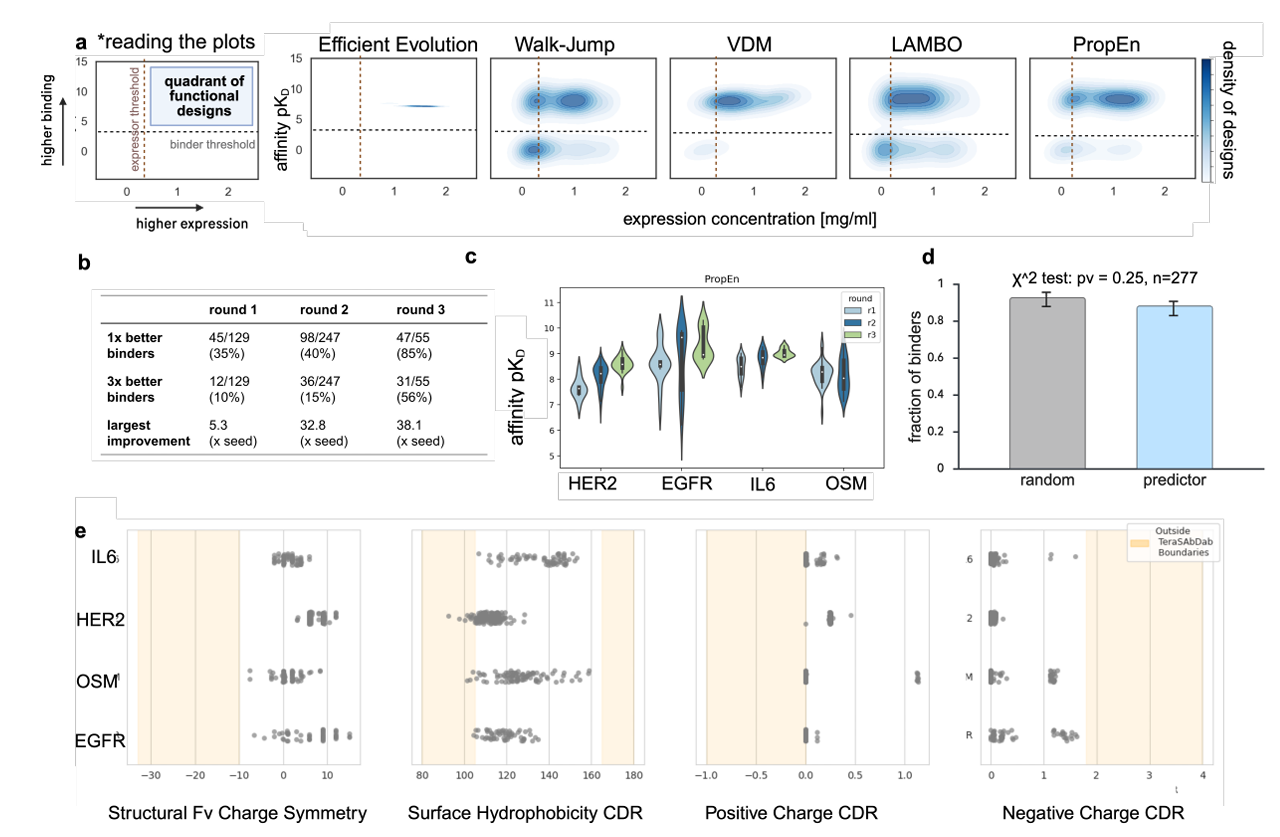
11개의 seed 항체, 4개의 서로 다른 항원 타깃에 대해 PropEn이 실제 실험에서 어떤 성능을 보였는지 요약
(a) Baseline과의 비교 – expression vs affinity
패널 (a)는 PropEn과 여러 baseline 기법(Efficient Evolution, Walk-Jump, VDM, LAMBO 등)을 발현량(expression; yield)과 결합 친화도(binding affinity) 기준으로 비교
첫 패널에서 정의: 설계된 항체가 기능적(functional)이라고 판단되려면 미리 정한 affinity threshold와 expression threshold를 모두 넘겨야 함
구체적으로 pKD < 5 인 경우는 non-binder(결합 못 하는 수준)로 간주
여기서 pKD = −log10(KD), KD는 dissociation constant
KD가 작을수록 결합이 잘 되고, pKD는 클수록 좋음
결과: PropEn으로 생성한 설계들의 분포가 그래프의 오른쪽 위 구석에 가장 많이 모여 있음
→ 높은 affinity + 높은 expression을 동시에 달성하는 설계가 많다는 뜻
(b, c) 여러 라운드의 lead optimization
패널 (b)와 (c)는 PropEn을 사용해 여러 라운드의 lead optimization을 수행했을 때 결과가 어떻게 개선되는지를 보여줌
여러 번 실험·설계를 반복할수록 여러 타깃에 걸쳐 lead가 점점 개선됨
즉, PropEn이 단발성 설계가 아니라, 반복적으로 품질을 끌어올리는 도구라는 걸 실험으로 확인
(d) A/B 테스트 – predictor 사용 여부의 영향
패널 (d)는 실험에 보낼 설계를 고를 때 “predictor”를 쓰는 게 의미가 있는지를 테스트
A/B 테스트 결과: predictor를 써서 후보를 고른 실험과,
그냥 고른 실험 간에 유의미한 차이가 없다고 보고
PropEn 자체가 이미 좋은 설계 분포를 만들어주기 때문에, 그 위에서 추가로 predictor를 써도 실험 결과가 크게 달라지지 않는다는 의미
(e) Developability – TAP로 본 치료제 적합성
패널 (e)는 PropEn 설계들의 developability(개발 가능성) 특성을 평가
사용한 도구: Therapeutic Antibody Profiler (TAP)
-> 여러 구조·물성 기준으로, 항체가 실제 치료제로 적합한지 평가해주는 툴
결과: PropEn 설계 항체들의 96%가 TAP가 정의한 “유효한 치료제 범위(valid therapeutic ranges)” 안에 들어감
즉, PropEn은 단지 affinity 좋은 “괴물 항체”를 만드는 게 아니라, 실제로 약으로 쓰기 좋은 프로파일을 가진 항체를 만들어낸다는 것을 보여줌
✅ 6. Iterative refinement
PropEn의 encoder-decoder 구조 덕분에, 같은 모델을 여러 번 돌려서 하나의 lead를 점점 더 개선하는 게 가능함
구체적으로, 초기 seed 항체를 넣어서 PropEn이 여러 mutant를 만듦
그중 실험 또는 예측 상 좋은 것들을 다시 PropEn의 입력으로 넣음
이 과정을 반복하면 최적화 궤적(trajectory)를 따라 lead가 점점 더 좋은 property로 이동
논문은 이렇게 iterative한 사용 방식이 기존 generative 모델들(예: diffusion, LLM 등)보다 고품질 + 기능적으로 의미 있는 설계를 얻는 데 더 효과적이라고 주장
✅ Result
✅ 2.1 PropEn은 여러 타깃에서 in-vitro로 기능성 항체를 만든다
PropEn을 실제 실험(in-vitro)으로 검증
타깃 항원은 4개: IL-6, HER2, EGFR, OSM
각각의 타깃에 대해 총 11개의 seed 항체(초기 리드 분자)를 사용
목표: 결합 친화도(affinity)를 올리면서 발현량(yield)를 유지하거나 더 좋게 만드는 것
결합 친화도 측정은 SPR (Surface Plasmon Resonance) 실험으로 함
성능 결과: PropEn은 모든 타깃에서 리드 항체들을 성공적으로 최적화함
94.6% binding rate
→ 설계된 항체들 중 약 94.6%가 타깃에 의미 있게 결합
98% expression rate
→ 98%가 발현이 잘 되는 수준을 넘김
비교 대상 SOTA 모델들보다 성능이 좋음
VDM: 86%
WJS: 62.9%
LAMBO: 44.2%
→ PropEn이 binding, expression 모두에서 우월
왜 이렇게 잘 되나?
PropEn은 친화도 + 발현량을 동시에 최적화하도록 설계되어 있음
그래서 설계된 항체들이 기능적으로 유효한 분포(functionally relevant distribution) 안에 남아 있음
즉 “이론상 이상한 서열”이 아니라, 실제로 잘 발현되고 결합할 가능성이 높은 구역만 탐색
PropEn은 로컬 sequence space(seed 주변의 작은 변이 공간)를 탐색하면서 새로운(novel) 변이이지만 구조적으로는 말이 되는(viable) 변이들을 찾아냄
덕분에 분자 기능(affinity 등)을 향상시키면서도 여러 developability 특성(발현, 안정성, liabilities 등)을 함께 유지할 수 있음
✅ 2.2 PropEn은 다른 ML 방법보다 “데이터 효율”이 훨씬 좋다
PropEn이 데이터를 얼마나 적게 쓰면서도 성능을 내는지 기준으로 비교
비교 대상
EffEvo (Efficient Evolution): 수백만 개 단위의 protein sequence들로 학습한 LLM 기반 방법
WJS, LAMBO: OAS(항체 레퍼토리 DB)와 내부 데이터셋을 활용
VDM: 10x NGS(대용량 시퀀싱) 데이터로 학습
→ 다들 엄청 큰 데이터셋을 써서 학습한 baseline들
PropEn 쪽 데이터 조건
PropEn은 각 타깃마다 “수백 개(several hundreds)” 정도의 서열만 가지고 학습
그러니까 데이터 양으로 치면 baseline들에 비해 한참 적은 수준
그럼에도 불구하고 PropEn은 affinity maturation(친화도 향상) 측면에서
모든 baseline을 이김
즉, PropEn은 적은 training data로도 높은 품질의 설계를 만들어내는, 매우 sample-efficient한 방법임
✅ 2.3 PropEn은 외부 guidance(예측 모델)에 의존하지 않는다
PropEn은 설계한 항체들이 다시 여러 predictor(affinity 예측 모델 등)를 거쳐 필터링된 뒤 실제 실험으로 가는 구조였음
그래서 “혹시 PropEn이 잘 되는 이유가, 이 외부 predictor 덕분은 아닌가?” 하는 의문을 검증해야 했음
동일한 파이프라인 안에서 A/B 테스트를 수행
PropEn이 만들어낸 설계들을 두 그룹으로 나눔:
Group A: predictor들이 계산한
예측 affinity pKD
binding probability
방법별 ranking score
등을 사용해서 선별
Group B: affinity prediction 값을 랜덤하게 섞어(shuffled) 사용하고 global selection만 적용
→ 즉, 사실상 의미 없는(random) guidance를 사용한 그룹
결과: 차이가 없음 (Figure 2-d)
Group A: 약 94.2% binders
Group B: 약 95.2% binders
얼핏 보면 B가 조금 더 높은 것 같지만, 카이제곱 검정(χ² test) 결과: p = 1.0 → 통계적으로 차이가 전혀 없다고 봐도 무방한 수준
A와 B의 차이는 그냥 우연한 변동(random variation) 범위 안
어떤 그룹도 다른 그룹보다 “진짜로 더 잘한다”고 볼 수 없음
결론: PropEn의 성능은 외부 predictor가 가이드해줘서 나온 게 아님
PropEn 내부 모델 자체에 “어떤 변이가 좋아지는 방향인지”에 대한 guidance가 이미 내장되어 있음
✅ 2.4 PropEn은 복잡한 데이터셋에서 “희귀하지만 의미 있는 변이”를 찾아내고 강화(enrich)한다
PropEn이 어떤 식으로 의미 있는 mutation을 골라내는지
분석 대상: N032 anti-EGFR seed
PropEn이 제안한 변이들 중, N032 anti-EGFR seed 항체를 변형한 설계들을 집중 분석
특히 첫 번째 design round에서 나온 결과만 모음
이 라운드에서는 seed 주변에 사용 가능한 데이터가 매우 적었다는 점이 중요
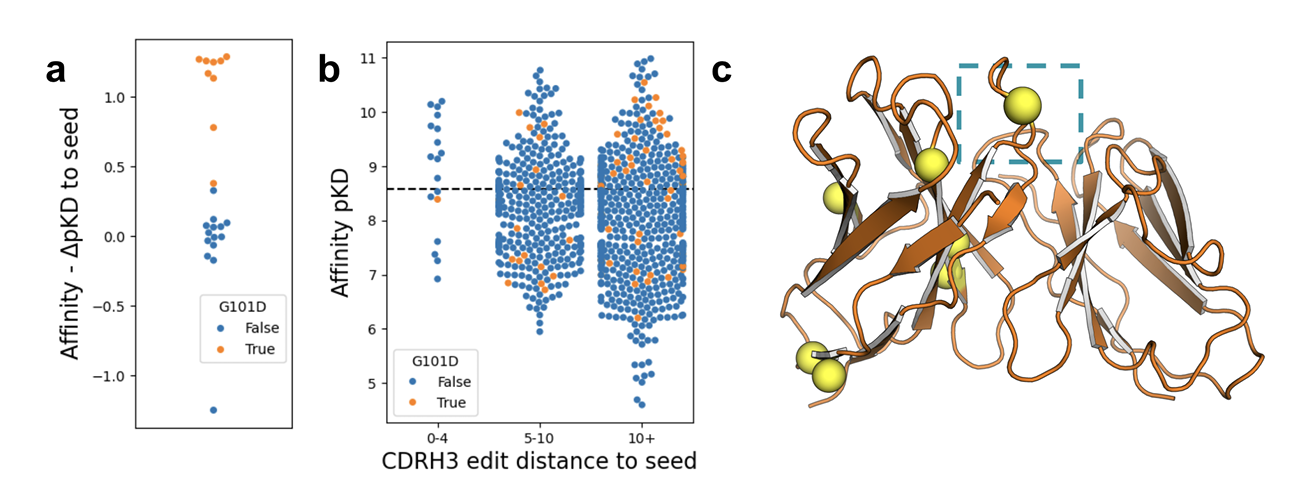
발견 1: 거의 모든 좋은 설계에 공통으로 들어간 G101D
binding affinity가 개선된 설계들은 거의 예외 없이 CDRH3 영역의 G101D 변이를 포함하고 있었음
(Figure 3-a)
이게 너무 눈에 띄어서, “이 G101D라는 변이가 원래 학습 데이터에는 어떻게 들어있었지?”를 조사하게 됨
학습 데이터 확인
PropEn을 학습시킬 때 사용했던 데이터셋:
EGFR를 타깃으로 하는 다양한 naive / affinity-matured clone들이 포함
총 921개의 역사적 측정값
서로 다른 epitope(항원 부위)를 타깃으로 한 clone들도 섞여 있었을 수 있음
중요한 점: 이 데이터는 학습 전에 어떤 전처리도 하지 않았음
조사 결과: G101D 변이가 들어간 clone들이 데이터셋에 실제로 존재하고 있었음
하지만 이 변이가 binding affinity에 미치는 영향은 비선형적(non-linear)이었음
즉, 어떤 clone에서는 G101D가 affinity를 올리는 역할을 했고
다른 clone에서는 오히려 affinity를 떨어뜨렸다 (Figure 3-b)
그래서 전체 데이터만 보면 “좋은 변이로 enrichment”되어 있지 않았음
게다가, G101D는 주로 seed와 sequence 거리가 먼 clone들에서 나타났음
특히 CDRH3 부분에서 seed와 edit distance가 0~4처럼 가까운 clone들에서는 G101D가 존재해도 affinity에 거의 영향을 주지 않았음 (Figure 3-b)
이게 의미하는 바
데이터 전체만 보면 G101D는 좋을 때도 있고, 나쁠 때도 있고, 별 영향이 없을 때도 있는 변이
그런데도 PropEn은 seed N032 주변의 로컬 sequence space에서
“G101D가 들어가면 affinity가 확실히 좋아지는 조합”을 찾아낸 것
논문이 강조하는 포인트:
PropEn의 sequence matching 전략(비슷한 서열끼리 매칭), 모델 구조 설계 덕분에,
이렇게 복잡하고 전처리 안 된(non-curated) 데이터에서도 겉으로 드러나지 않는(non-obvious) 유리한 변이를 발견하고, 그 변이를 가진 설계를 많이 만들어(enrich) 낼 수 있음
잠재적인 단점: 프레임워크 변이가 너무 많아질 수 있음
PropEn은 때로 서로 거리 먼 clone들끼리 pair를 만들 수 있음
이 경우, CDR에서 의미 있는 변이와 함께 프레임워크 영역에 불필요한 변이가 많이 딸려 들어올 수 있음
실제 예시: N032 seed에 대해 PropEn이 찾은 최고 성능 변이체는
총 7개의 edit을 가지고 있었는데, 그 중 6개가 framework 영역,
1개만 CDRH3 영역의 G101D였음 (Figure 3-c)
분석에 따르면 affinity에 직접적이고 재현 가능한 효과가 있는 변이는 사실상 G101D 하나뿐이었음
아마 EGFR 항원과의 추가 상호작용을 만들어 주었기 때문으로 추정
다행히도, 이런 “프레임워크 과도 변이” 문제는 데이터셋 준비 단계에서 pairing 전략을 더 신중히 짜거나 실험 전에 post-processing 단계에서 프레임워크 변이를 정리하는 방식으로
상당 부분 해결할 수 있다고 언급
✅ 2.5 PropEn은 다양한 developability 속성을 최적화한다
이제는 단순 affinity 말고, 여러 developability property에 대해 in-silico 평가
PropEn의 최적화 능력을 검사하기 위해, in-silico 실험을 수행
비교 대상: 여러 state-of-the-art 방법들
평가 대상 property:
항체의 sequence 기반, structure 기반 특성들
(hydrophobicity, charge, liabilities 등)
각 속성에 대해의 목표:
held-out test point(보지 않은 테스트 항체)에 대해
Δproperty(parent − mutant)를 최대화하는 것
쉽게 말하면, 같은 lead(부모) 항체를 기준으로 PropEn이 만든 mutant가 얼마나 property를 개선했는지 점수로 봄
모델 구성: 각 property마다 별도의 PropEn 모델을 학습
결과
모든 property 평가에서 PropEn이 per-point Δproperty가 가장 큼
PropEn이 다양한 developability 측면을 고려하면서도, 기능적으로 향상된 설계를 만드는 능력이 있다는 뜻
Table 1에 각 방법별로 속성 개선된 설계 비율(%)을 정리
PropEn은 모든 속성에서 다른 baseline보다 개선 비율이 가장 높고, 50–80% 수준의 improvement rate를 달성한다고 정리
Table 1 – in-silico property 개선 비율 비교
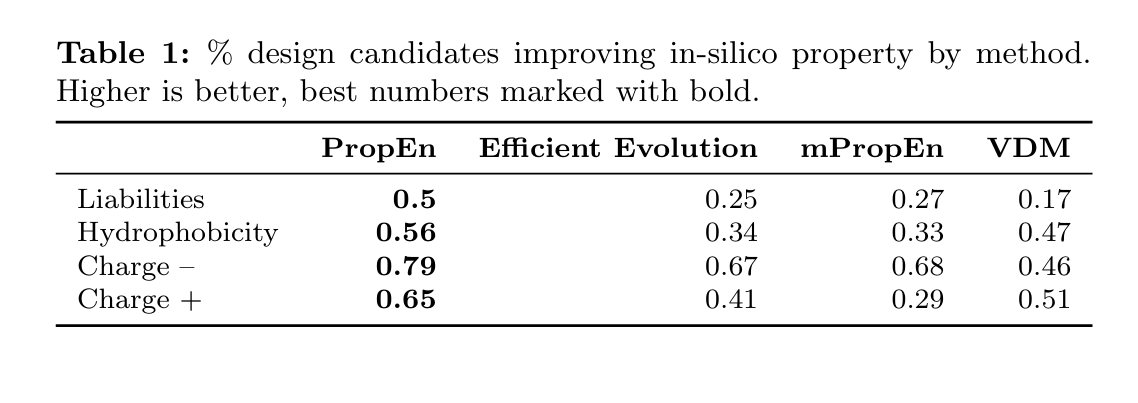
Liabilities: 화학적/물리적 문제를 일으킬 수 있는 “위험 요소” (예: aggregation motif 등)
-> 값이 낮을수록 좋음
Hydrophobicity: 너무 소수성이 강하면 aggregation이나 낮은 용해도 문제
-> 역시 줄이는 방향으로 개선된 설계 비율
Charge − (음전하)
Charge + (양전하)
전하 분포도 developability에 중요한 요소라, 각각을 안전한 범위로 조정하는 것을 목표로 함
네 가지 property 모두에서 PropEn이 최고값(볼드)
✅ 2.6 mPropEn은 다중 속성을 동시에 최적화한다
이제 단일 property 각각이 아니라, 여러 property를 한꺼번에 최적화하는 세팅
PropEn에 다변량 목적 함수(multivariate objective function)를 도입
이 함수는 설계된 항체의
hydrophobicity
charge(양전하·음전하)
liability profile
을 종합해서 하나의 ranking score로 만듦
multivariate 점수 계산에는 BOtied [19]라는 기법을 사용
다목적 베이지안 최적화류 방법으로 보이며,
이를 활용해 multi-objective PropEn (mPropEn)을 돌림
mPropEn은 동기적으로 gradient를 계산해야 하는 differentiable objective만 가능한 기존 최적화 방법들과 달리,
미분 가능하지 않은 복잡한 objective들(여러 계산 property 조합)도 다룰 수 있다는 점을 강조
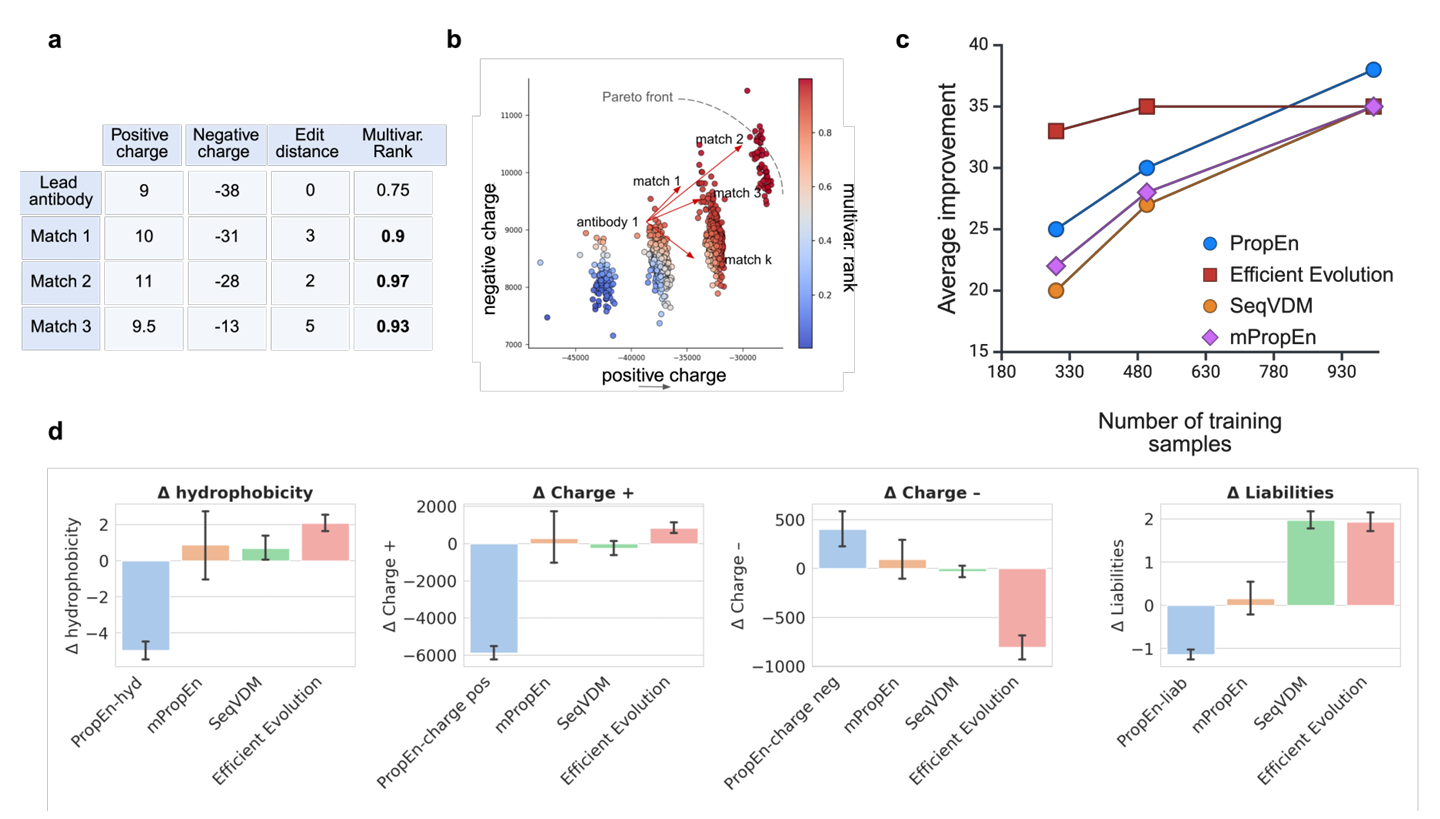
Figure 4 – multi-property 최적화 & in-silico 평가
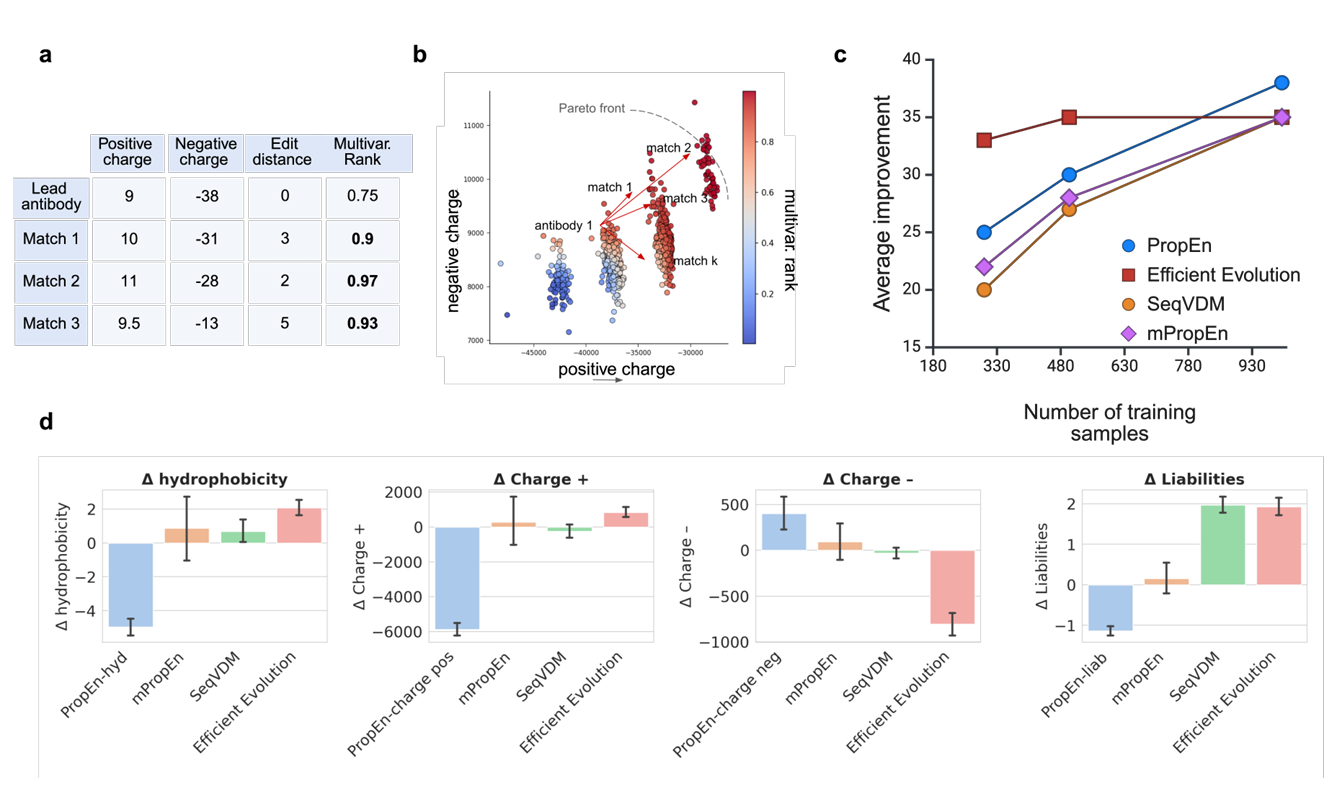
Fig. 4a – multivariate ranking score가 matching에 쓰이는 예
하나의 Lead 항체에 대해, 각 Match(매칭되는 다른 항체)는 하나 이상 property가 lead보다 같거나 좋음
그런 match일수록 전체 multivariate rank가 높게 매겨짐
이 과정을 통해 매칭 단계에서 이미 다중 속성을 동시에 고려하게 됨
Fig. 4b – multivariate rank와 Pareto front의 관계
multivariate rank score가 항체들을 올바른 순서로 정렬해 준다는 걸 보여줌
Pareto front에 가까운 설계일수록 색깔이 더 진한 붉은색(darker red)으로 표시됨
즉, 이 점수가 “Pareto front에 얼마나 가까운지”를 잘 반영하고 있음을 시각적으로 보여줌
Fig. 4c – 데이터 효율 (sample efficiency) 비교
PropEn과 mPropEn의 sample efficiency를 Efficient Evolution, VDM 같은 baseline들과 비교
같은 수의 training sample을 쓸 때, PropEn/mPropEn이 평균 property 개선량(average improvement)에서 항상 더 좋은 성능을 내는 트렌드를 보여줌
Fig. 4d – 계산 속성들에 대한 방법 비교
각 방법이
hydrophobicity 감소
양전하(Charge +) 감소
음전하(Charge −) 조절
liability 개수 감소
를 얼마나 잘 이루는지 비교
결과:
PropEn은 VDM(확산 기반)과 Efficient Evolution(단백질 LLM 기반)보다
각 lead-mutant 쌍에서의 property 변화(Δ property) 기준으로 항상 우수
mPropEn은 여러 목표(property) 사이의 균형(balance)를 잘 유지함
즉, 어느 한 property만 과하게 희생시키지 않고, 여러 property를 동시에 적절히 trade-off
Pareto front 및 최종 결론
다중 속성 최적화에서는, 모든 속성을 무한정 좋게 만들 수 없고
서로 trade-off가 발생하므로, Pareto front에 최대한 가까운 설계를 찾는 것이 중요
✅ Discussion
✅ 3.1 PropEn을 설계한 다섯 가지 목표
(1) Target-agnostic
특정 항원(HER2, EGFR 등)에만 맞는 모델이 아니라 어떤 리드 항체에 대해서도 다양하면서도 최적화된 설계 라이브러리를 제안할 수 있어야 함
즉, “특정 타깃 전용이 아니라, 타깃에 독립적인 일반적인 도구”가 되게 하려는 목표
(2) Property-agnostic
친화도, 발현량, 안정성, 용해도 등 어떤 특성(property)이든
관심 있는 항목에 맞춰 적용할 수 있어야 함
즉, “이 모델은 affinity 전용이다”가 아니라, 관심 있는 property를 바꿔도 그대로 쓸 수 있는 프레임워크를 지향
(3) Novelty in property values
단순히 기존 데이터와 비슷한 property를 재현하는 게 아니라 out-of-distribution, 새롭고 더 좋은 property 값을 가진 항체도 설계해야 함
(4) 동시 multi-property 최적화
여러 특성(affinity, expression, liabilities 등)을 동시에 최적화할 수 있어야 함
한 가지 property만 올리고 나머지는 망가뜨리는 방식이 아니라
trade-off를 고려한 동시 최적화가 목표
(5) No-guidance required
외부의 복잡한 guidance(예측 모델, 별도의 스코어링 네트워크 등)에 의존하지 않는 방법이어야 함
저자들은 PropEn이 이 다섯 가지 목표를 실제로 달성한다는 것을 확인했으며, 그 핵심 이유가 pair matching 절차 때문이라고 강조
✅ 3.2 Generalizable + Efficient 한 이유
PropEn은 일반화 가능(generalizable) 하고, 효율적(efficient) 이라서,
각 (항원) 타깃마다 아주 작은 데이터셋만 있어도,
단 한 번의 wet-lab 라운드에서 binders를 뽑아낼 수 있다고 주장
데이터 양과 질에 대한 설명
각 타깃당 가진 데이터는 “몇백 개 수준의 포인트” 뿐
이 데이터들은 동일한 실험실에서 새로 만든 게 아니라,
조직 내 여러 팀들이 과거에 수행했던 실험에서 모은 것
→ 실험 조건도 다르고, 항체 종류도 제각각인 이질적인 데이터
그럼에도 불구하고, in-silico 실험과 baseline 비교에서,
high-throughput mutagenesis 데이터로부터 샘플을 사용했을 때도 PropEn이 우위
in-silico, 물리기반 property score의 역할
in-silico, physics-based property score 들이 존재한다는 점 덕분에,
저자들은 PropEn을 대상으로
target-agnostic / property-agnostic 성질,
multi-property optimization (mPropEn) 능력을
체계적으로 평가할 수 있는 실험 셋업을 만들 수 있었음
이 in-silico 실험들에서 본 PropEn의 장점:
absolute property improvement: seed와 디자인 사이의 Δ(property)가 가장 큼
controlled generation: 아무 서열이나 막 뽑는 게 아니라, 원하는 분포 안에서 안정적으로 샘플을 생성
LLM 기반 baseline과의 대비: Language Model 기반 baseline들은 리드(lead) 주변에서 유효한(valid) 서열을 잘 제안하지만,
guidance가 없기 때문에, 실제 관심 property(예: binding affinity)는 개선되지 않는다고 함
Figure 2a에서 Efficient Evolution은 binding gain이 전혀 없는 것으로 나타남
WJS도 비슷한 패턴
반면 LAMBO 같은 guided baseline은 탐색(exploration)을 강하게 해서 binding을 크게 올리긴 하지만, 그 대가로 발현이 잘 안 되는 non-valid 서열들을 많이 만듦
PropEn은 매칭된 pair들을 항상 서로 가까운 서열로 유지하면서
그 안에서 property만 좋아지는 위치들을 학습
즉, 모든 면에서 seed와 비슷하지만, 딱 필요한 몇 개 위치만 바뀌어 property가 좋아지는 “faithful mutations”만 배움
이런 식으로 작고 정밀한 neighborhood를 학습하면, 항상 valid한 분자(발현·안정성 면에서 문제 없는 항체)를 90% 확률로 제안할 수 있다고 주장
✅ Methods
✅ 4.1 PropEn 개요
PropEn은 matched-pair encoder–decoder framework
적은 양의 sequence–property 측정 데이터를 “생성 최적화기(generative optimiser)”로 변환하는 것
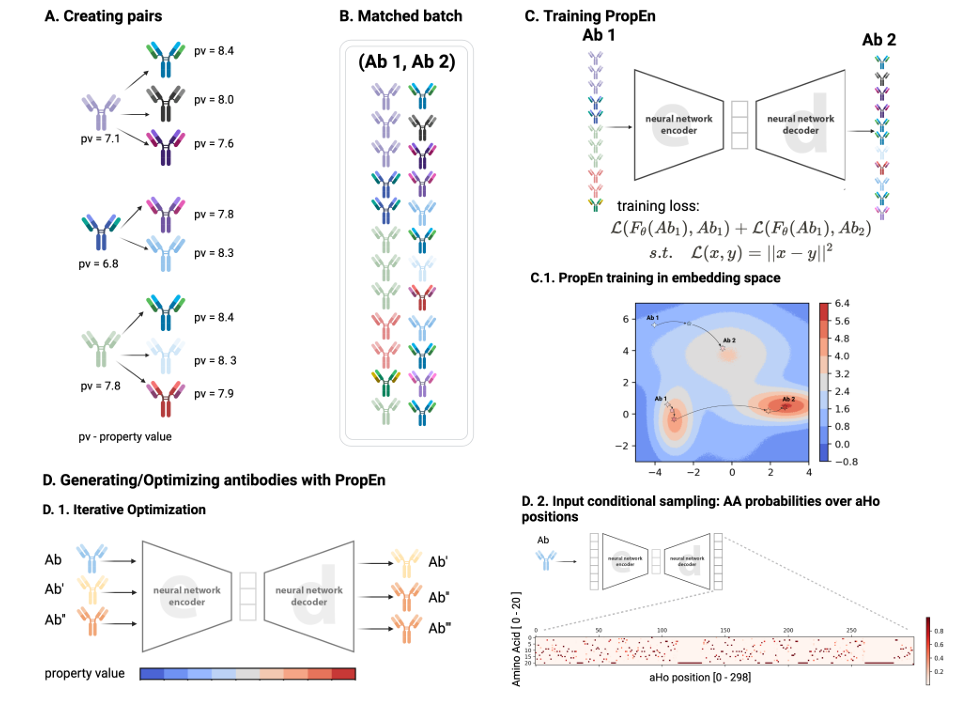
(a) Pair 만들기 – counterfactual neighbour 찾기
training set의 각 항체에 대해, counterfactual neighbour를 하나 찾음
조건: 그 neighbour 서열은 원래 서열과 최대 δₓ 개 포인트 변이까지만 차이가 나고, 관심 property 값이 더 큼 (Δy ≥ δᵧ).
이렇게 해서, “거의 비슷하지만 property가 더 좋은” 쌍을 구성
(b) Matched batch 구성
위에서 만든 쌍들 (Ab1,Ab2)이 학습에 쓰이는 minibatch를 이룸
Ab1: lower-property 서열
Ab2: higher-property 서열
(c) PropEn 모델 – auto-encoder로 학습
PropEn은 lightweight ResNet auto-encoder 구조
입력: property가 낮은 서열 Ab1.
목표: property가 높은 파트너 Ab2를 재구성(reconstruct) 하도록 학습
이 과정이 앞에서 설명한 paired reconstruction loss
(self + counterfactual reconstruction)와 연결
(d) 학습 후 새로운 설계 생성
학습이 끝난 뒤에는 두 가지 방식으로 새 서열을 만듦
(d₁) Iterative re-feeding (반복 최적화)
decoder 출력된 서열을 다시 encoder/decoder에 넣는 과정을 반복
수렴할 때까지 혹은 edit budget을 채울 때까지 반복 → Section 4의 기본 모드
(d₂) Sampling from final-layer categorical distribution
decoder 마지막 층의 categorical 분포에서 사용자가 설정한 temperature로 샘플링
temperature를 조절해서 탐색(exploration) vs 착취(exploitation)의 균형을 조절
기존 방식: unconditional generator (그냥 서열을 만드는 생성기)
별도로 학습된 surrogate predictor (property 예측 모델)
→ 이 둘을 조합해 최적화
문제점: surrogate predictor가 완벽하지 않아서 adversarial false positives(예측은 좋다고 나오지만 실제로는 별로인 경우)에 취약
PropEn 방식: matching을 통해 “어떤 변이가 property를 개선하는지”라는 improve 신호를 직접 생성 모델 안에 통합
그래서 별도의 property predictor 없이도 최적화가 가능
✅ 4.2 Matching strategy


왜 이런 매칭을 쓰는가?
이 매칭은 인과 추론(causal inference)에서의
nearest-neighbor matching 아이디어에서 영감을 받음
결과적으로 두 그룹을 만듦
treatment group: property가 더 좋은 항체 (higher property)
control group: property가 낮은 항체 (lower property)
두 그룹은 아주 국소적인 local edits만 다르면서 property는 의미 있게 차이나도록 구성

✅ 4.3 Model architecture and training
네트워크 구조
Encoder: 3개의 블록으로 이루어진 1D ResNet
파라미터 수: 3.4M
Decoder: encoder와 가중치를 공유(tied-weights)하는 decoder
입력: AHo 넘버링으로 정렬된(one-hot AHo-aligned) 서열
처리: 이 one-hot 서열을 128차원의 latent space로 투영
ReLU activation, layer normalization을 전체 네트워크에서 사용
학습 objective

이 두 번째 항 덕분에 latent manifold가 property gradient 방향으로 순서(order) 잡히게 됨
이 현상은 t-SNE로 latent를 시각화하고 y값으로 색칠해서 확인함
✅ 4.4 Sequence generation
실제 inference 때, PropEn은 seed 항체 𝑥(0)를 입력으로 받고 두 가지 생성 모드를 제공
1) Iterative optimisation (기본 모드)
반복적으로 decode → encode
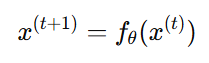
멈추는 조건:
새로 생성된 서열과 이전 서열 사이의 편집 거리
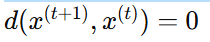
더 이상 변이가 안 생길 때 혹은 사용자가 정한 edit budget(허용 변이 횟수)이 소진될 때
경험적으로: property 값은 보통 5번 이내의 iteration에서 plateau(더는 크게 증가하지 않음) 한다고 함
2) Stochastic sampling (확률적 샘플링 모드)
각 위치의 pre-softmax logit을 하나의 categorical 분포로 해석
temperature 𝜏∈[0.1, 1.5]를 사용해:
𝜏가 낮을수록 → 더 보수적인 샘플 (원래 서열과 유사)
𝜏가 높을수록 → 다양한 서열
이렇게 하면 seed 𝑥(0) 주변에서 다양한 변이체(mutant)를 생성할 수 있음
필요하다면, 생성된 샘플에 대해 한두 번의 iterative optimization을 더 돌려서 property를 조금 더 끌어올리는 hybrid 모드도 가능
✅ 4.5 Multi-property extension – mPropEn
실제 약물 후보는 여러 developability 조건을 동시에 만족해야 함
이를 위해 PropEn의 matching을 벡터형 property로 확장
스칼라 𝑦 대신, 여러 property (𝑦1,...,𝑦𝑚)를 multivariate rank score 𝑠 로 합침

이렇게 학습된 mPropEn은 in-silico와 in-vitro 모두에서 Pareto optimality(다중 목표 최적해)에 가까운 설계를 유지
✅ Baselines
대규모 language model 및 diffusion model들을 baseline
EffEvo: 일반적인 단백질 언어 모델 ESM-2를 활용
항원 정보, 특이성, 구조 정보를 전혀 쓰지 않고, 진화적으로 그럴듯한 변이를 제안하는 방식
원래 연구에서, 딱 몇 개의 변이만 실험해도 두 번의 실험 라운드 안에 높은 affinity 향상을 달성했다고 보고
WJS: score-based + energy-based 모델을 smoothed discrete protein sequence manifold 위에 학습
Langevin MCMC로 noisy sequence space를 “걷고(walk)” 다니다가, one-step denoising을 통해 정확한 서열로 “점프(jump)”하는 구조
장점: non-autoregressive 생성이라 빠르고, 다양한 고품질 항체 서열을 생성 가능
VDM: latent-space diffusion model
입력 표현: one-hot AHo representation
변형:
guidance 없는 기본형 (VDM)
ILVR-guided 버전 (VDM-G)
LAMBO-2: masked-language 모델 + diffusion을 결합한 하이브리드 아키텍처
discrete diffusion model에 대한 gradient-based guidance를 통해
바로 sequence space에서 단백질을 설계
여기에 LaMBO-2는 edit-based 제약조건을 가진 multi-objective Bayesian optimisation을 얹은 버전
In-silico 실험: 모든 baseline을 동일한 컴퓨터 연산 자원 아래에서 validation 성능이 최대가 되도록 튜닝
In-vitro 실험: 각 baseline 논문의 저자들이 제안한 최적 설정으로 실행
각 모델은 서로 다른 아키텍처와 데이터셋을 사용하므로, 비용–효율성 trade-off도 제각각
특히 PropEn은 이들 중 모델 크기가 가장 작고, 필요한 데이터 양도 가장 적다고 강조
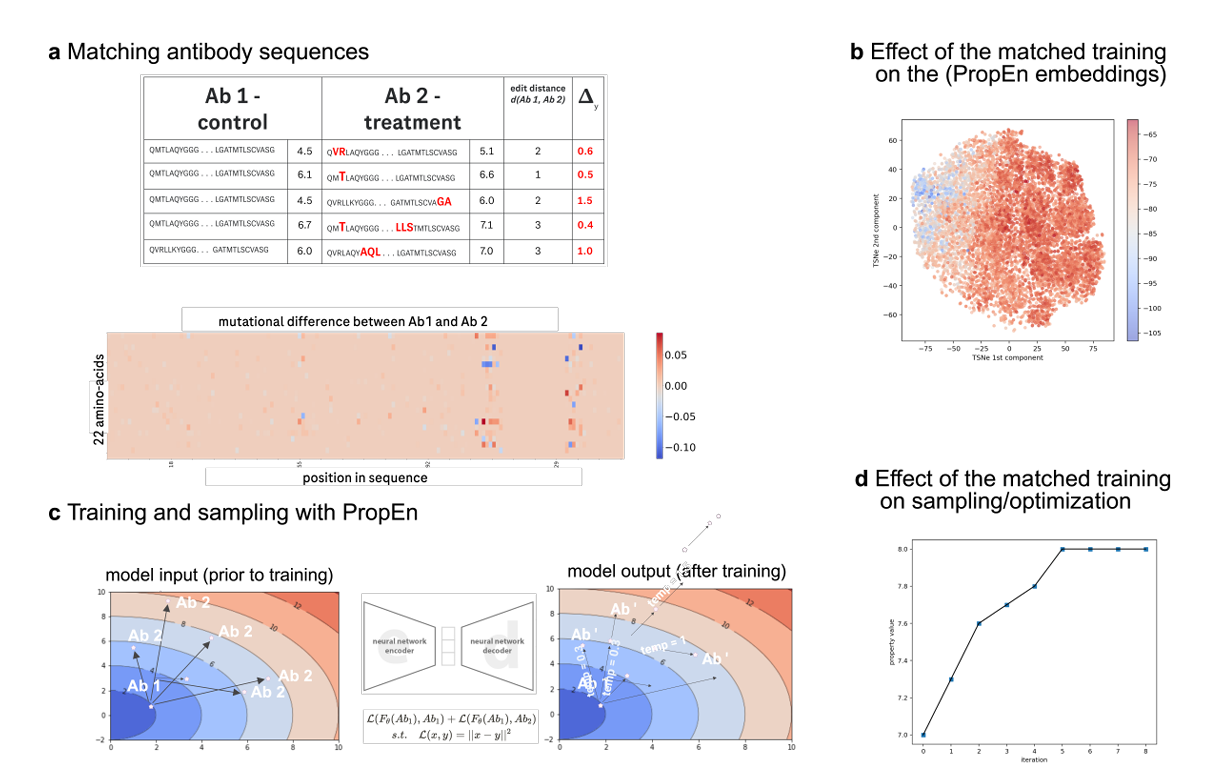
Fig. 5 – Matched training을 통한 property 향상 메커니즘
a. Matching antibody sequences
항체들이 서열 유사도(낮은 edit distance) 기준으로 control 그룹(낮은 property)과 treatment 그룹(높은 property) 쌍으로 묶임
즉, 거의 비슷하지만 property가 더 좋은/나쁜 쌍을 만드는 장면
b. Paired-loss로 continuous property landscape 학습
이런 쌍들이 PropEn의 paired loss를 학습하는 데 사용됨
목적: 연속적인 property landscape를 모델링하고, 그 위에서 단계적인(stepwise) antibody 개선 경로를 제안할 수 있게 만드는 것
c. t-SNE 시각화 – 학습된 단조 gradient
control 그룹(낮은 property)의 항체들을 PropEn의 embedding 공간에서 t-SNE로 시각화
각 포인트는 integral surface hydrophobicity(표면 소수성 통합값)을 색으로 표현
결과: 색 변화가 단조(monotonic) gradient를 이루는 모습이 보임
→ latent 공간이 실제 property 값과 정렬되었음을 의미.
d. Implicit gradient를 따라가는 iterative sampling
이 implicit gradient를 따라 반복적으로 샘플링하면 property가 차례로 향상되는 서열들을 얻을 수 있음
-> iterative optimisation 모드에서 설명한 것과 연결
✅ Datasets
In-silico developability properties

1) Hydrophobicity
Description: 분석 대상(analyte)이 수용성 환경이 아닌(non-aqueous) 환경을 선호하는 정도
이 특성은
약동학(pharmacokinetics),
효능(efficacy),
투여 간격(dose intervals),
투여 경로(route of administration)
등 여러 임상적 요소에 영향을 줄 수 있음
과도한 소수성은 aggregation / 용해도 문제를 일으키므로 줄이는 것이 목표
2) Positive & negative charge
Description: Fab 전체에 대한 surface patch integral
즉, 표면 전하 분포를 구조를 기반으로 통합한 값
전하가 너무 크거나 불균형하면 비특이적 결합, 안정성 문제 등을 야기할 수 있으므로, 안전한 범위로 줄이는 것이 목적
3) Liabilities
Description: aggregation motif, deamidation site 등 “Liability motif”들을 문헌에서 수집한 리스트를 기반으로 함
목표는 이 liability motif의 개수를 줄이는 것
motif마다 중요도(priority)를 매기는데, 1이 가장 중요(예: CDR 주변의 W 등)
CDR 주변 전반에 걸친 이러한 motif들을 카운트하고 관리
여러 종류의 liability를 따로 세기기 때문에 3차원으로 표현
4) Multi-property score
Description: 위 특성들을 포함한 모든 property에 대한 성능을 종합해서 Pareto front 기준으로 얼마나 좋은지 나타내는 점수
In-silico data
in-silico 실험은 yeast display library의 부분집합을 사용
이 라이브러리는 parental clone 4D5의 scFv(single-chain fragment variable)
4D5는 치료용 항체 Trastuzumab에서 유래했고, 인간 HER2에 결합하는 항체
기능을 유지하면서도 binding에 중요한 위치를 잘 탐색하기 위해
두 개의 CDR loop만을 변이 대상으로 선택: CDRH3, CDRL3
각 항체마다 9개의 아미노산 위치가 변이될 수 있음
4D5 라이브러리는 HER2 binding strength에 따라 세 그룹:
high-binding
low-binding
non-binding
이 중에서, 저자들은 HER2에 high binding을 보이는 population만 선텍
그리고 low data regime을 흉내 내기 위해 이 binder들 중 10%만 샘플링 → 11,556개 항체
각 라이브러리 샘플에 대해 6개의 in-silico developability property를 계산
이 properties는 구조 + 서열 기반이지만, PropEn은 sequence 데이터만 사용하기 때문에, “sequence만 가지고도 이런 developability를 개선할 수 있는지”를 테스트하기 위한 설정
각 property는, Table B1에 나온 해당 논문/방법들을 바탕으로,
Python 구현으로 계산
구조는 EquiFold를 사용해 예측