✅ Abstract
-
주제와 중요성
T-cell immunogenicity(펩타이드가 T-cell 반응을 유도하는 능력)는 단백질 치료제와 백신의 안전성과 효능을 좌우하는 핵심 요소 -
현 상황의 한계
딥러닝이 in silico 예측에 유망하지만, 면역원성 데이터가 포괄적으로 부족한 것이 큰 난제 -
무엇을 제안했나 (TITANiAN)
다중 도메인 딥러닝 프레임워크이며, 적대적 도메인 적응(adversarial domain adaptation)을 이용해 다양한 면역학적 데이터 원천을 통합
통합되는 데이터는 MHC 제시(EL), pMHC 결합 친화도, TCR–pMHC 상호작용, T-cell 활성화, 유래 생물(출처) 정보 -
검증과 1차 목표 성능
엄격한 누수 통제(데이터 리키지 컨트롤) 벤치마크로 검증
미세조정된 주 작업(TITANiAN-IM)에서 특정 펩타이드-MHC 쌍의 T-cell 활성화 예측을 탁월하게 수행 -
눈에 띄는 확장 성과 – 항체 ADA 예측
MHC 입력 없이도 치료용 항체의 ADA(anti-drug antibody) 유발 가능성을 예측 -
왜 잘 되나 (성공 요인)
생물학적으로 정당화된 설계와 데이터 기반의 다중 도메인 사전학습 덕분
각 구성 요소의 기여도는 광범위한 어블레이션 연구로 추가 확인
-
적용 범위의 유연성
유연한 사전학습 아키텍처 덕분에 분자 결합 예측 등 더 넓은 면역원성 관련 과제도 지원 -
의의
일관되고 견고한 성능을 보이며, 더 안전하고 효과적인 백신·단백질 치료제 개발을 앞당길 잠재력을 지님
✅ Introduction
✅ 무엇을 다루나
T-cell epitope immunogenicity: 단백질에서 유래한 짧은 펩타이드가 T세포 면역반응을 유발하는 능력
이는 적응면역에서 보호적/병리적 반응 모두에 관여하며, 정확한 이해는 항암 면역치료 설계, 감염병 백신 개발, 단백질 치료제의 원치 않는 면역원성 예측·완화 전략에 직결
✅ 기존 접근의 한계
실험적 방법: 항원 단백질로부터 겹치게 잘린(오버래핑) 펩타이드를 대량 스크리닝하는 전통법은 비용과 시간이 많이 들고, 에피토프 경계와 면역원성의 강도를 정확히 특정하기 어려움
계산 예측(in silico): 효율적 대안이지만, 다수의 기존 모델은 MHC–펩타이드 결합이나 TCR–pMHC 상호작용처럼 T세포 인지 과정의 일부 요소만 분리해 다룸
->
이런 단편적 초점은 통합적 생물과정을 반영하지 못해 정확도와 일반화를 떨어뜨리고, 실제 면역원성 평가·치료 설계에 신뢰성 있는 도구로 쓰이기 어렵게 만듦
✅ 이 논문의 해법
이런 한계를 넘기 위해 TITANiAN을 제시
다양한 면역원성 관련 데이터를 통합하여 T세포 에피토프 면역원성을 좌우하는 복합 결정요인을 학습하도록 고안된 딥러닝 프레임워크
모듈러 멀티태스크 아키텍처를 사용해 MHC–펩타이드 상호작용, TCR–pMHC 결합, T-cell 활성화 등 여러 도메인의 정보를 함께 활용함으로써, 보다 포괄적이고 예측력 있는 모델을 구축한다는 것이 핵심 주장
✅ Result
✅ Model Architecture and Training Strategy
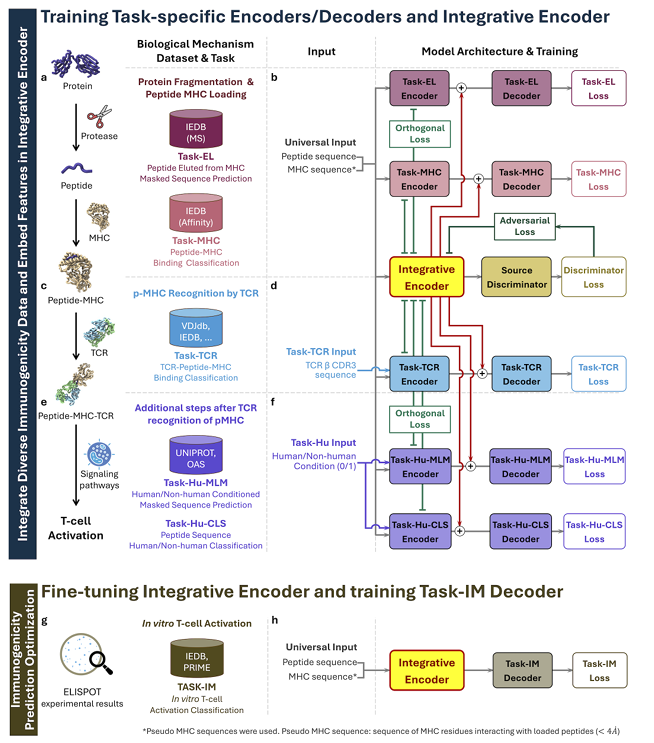
✅ 1) 모델 구조(무엇이 어떻게 연결되는가)
✅ 모듈형 멀티태스크 아키텍처:
공통 표현을 학습하는 Shared Integrative Encoder(공유 인코더)와, 각 도메인 신호를 뽑아내는 Task-specific Encoders/Decoders(도메인별 인코더/디코더)로 구성
공유 인코더는 “어느 데이터에서 왔는지 모르게” 만든(도메인-불변), 핵심 면역학 신호만 담고 = 도메인 불변(domain-invariant) 특징
태스크 인코더는 각 도메인(MHC-I/II, TCR 등)에 특화된 정보만 담도록 = 도메인 특이(domain-specific) 정보
강제로 역할을 분리
-> 이를 위해 직교 제약과 적대적 학습 사용
✅ 직교 제약과 적대적 학습
두 가지 표현을 분리(disentangle)하기 위해 직교 제약(orthogonality constraints)을 걸고,
적대적 손실(adversarial loss)과 소스 판별기(discriminator)로 공유 인코더가 데이터 출처를 구분 못 하도록(gradient reversal) 학습시켜
과적합(예: MHC-I/II 길이 차이)을 줄이고 핵심 면역원성 신호를 잡게 함
✅ 직교 제약(Orthogonality constraints): “역할 안 겹치게”
공유 표현 𝑒integrative 와 태스크 표현 𝑒task 사이의 내적(점곱)을 0에 가깝게 만드는 손실을 추가
→ 두 벡터가 수학적으로 직교(정보 상 중복 최소)하도록 유도해, 공유는 ‘공통 신호’, 태스크는 ‘특화 신호’만 담게 분리(disentangle)
같은 문서를 두 명이 요약할 때,
한 명(공유 인코더)은 “어느 출처든 통하는 핵심만”,
다른 한 명(태스크 인코더)은 “출처/도메인에 특화된 디테일만” 적게 하고,
겹치면 벌점 주는 느낌
✅ 적대적 손실 + 소스 판별기(Adversarial loss with discriminator): “출처 정보 지우기”
소스 판별기(작은 MLP)가 공유 표현 𝑒integrative만 보고 데이터 출처(EL, MHC-I/II, TCR 등)를 맞히려 함
Gradient Reversal Layer(GRL)로 역전된 기울기를 공유 인코더에 흘려서, 판별기가 잘 맞힐수록 공유 인코더는 ‘출처가 안 드러나게’ 표현을 바꾸도록 학습됨
결과적으로 공유 인코더는 도메인-불변 표현을 배우게 됨
✅ 왜 필요?
MHC-I 펩타이드는 주로 9–10mer, MHC-II는 더 김
모델이 “길이만 보고” 클래스를 구분하는 지름길에 과적합하면, 새로운 도메인/코호트로 가면 성능이 떨어짐
적대적 학습은 공유 인코더가 길이 같은 출처-특이 단서를 표현에 담지 못하게 만들어 일반화를 높여 줌
최종 훈련은 사전학습→미세조정의 2단계로 진행
연결 방식과 디코더: 필요 시 게이티드 어텐션(gated attention)으로 다중 입력을 통합하고, 공유 인코더/태스크 인코더의 임베딩을 연결(concatenate)해 간단한 MLP 디코더로 각 과제를 예측
✅ 2) 학습 전략(두 단계)
① 멀티태스크 사전학습(pre-training): 면역원성과 관련된 다양한 데이터셋을 동시에 학습해 넓은 생물학 지식을 통합
② 태스크 특화 미세조정(fine-tuning): 공유 인코더를 T-cell 활성화(Task-IM) 소규모 데이터로 미세조정하고, Task-IM 디코더를 함께 훈련해 최종 면역원성 점수를 출력
✅ 3) 사전학습 태스크와 사용 데이터(무엇을 얼마나 썼나)
EL (Eluted Ligand)
LC–MS로 MHC가 실제로 제시한 펩타이드를 모아놓은 데이터
논문에선 이를 이용해 제시·프로세싱 신호를 학습하는 사전학습 과제 Task-EL(I/II)로 씀
면역반응 초기 단계(항원 제시)를 대표
MHC (peptide–MHC binding)
펩타이드와 MHC 결합 친화도(강도) 측정치
사전학습 과제 Task-MHC(I/II)에서 결합 강도 예측을 통해 상호작용 정보를 학습
면역반응의 결합 단계를 반영
TCR (T-cell receptor)
TCR–pMHC 결합/인지 데이터
사전학습 과제 Task-TCR로, 제시/결합을 넘은 다운스트림 인지 단계의 신호를 학습(정제된 65,030 포인트 사용)
IM (Immunogenicity)
최종 T-cell 활성(면역원성) 라벨
ELISPOT 같은 기능성 실험으로 측정된 데이터를 모아 미세조정(Task-IM)에 사용
즉, 모델이 최종적으로 맞추려는 엔드포인트
IEDB, MHC BN, NCI, PRIME-2.0 등에서 모은 12,782건
EL(제시) → MHC(결합 강도) → TCR(인지) → IM(실제 T세포 활성)
= 항원 인식 연쇄의 초기 → 중간 → 최종 단계를 각각 대표하는 데이터셋/태스크들, 논문은 이들을 사전학습(EL/MHC/TCR) + 미세조정(IM)으로 엮어 성능을 끌어올림
Task-EL (I, II): 제시 펩타이드(EL, LC-MS) 마스킹 예측
IEDB EL 데이터 Class I 557,151 / Class II 206,055 엔트리 사용
-> BigMHC, NetMHCpan이 활용해 온 유형.
Task-MHC (I, II): pMHC 결합 친화도 예측
IEDB·MHCflurry 계열이 쓰는 포맷으로, Class I 129,973 / Class II 516,463 측정치.
Task-TCR: TCR–pMHC 결합(다운스트림 인지) 신호 학습.
IEDB·문헌에서 65,030 포인트 정제.
DLpTCR 등 상호작용 특화 방법이 놓치는 부분 보완.
Task-Hu-CLS / Task-Hu-MLM: Selfness(인간성) 학습.
OAS, UniProt 유래 인간/비인간 서열로
(i) 이진 분류(Hu-CLS), (ii) 종 조건부 MLM(Hu-MLM) 수행(아이디어는 Hu-mAb, AbNatiV, BioPhi 참고).
Task-IM(미세조정 데이터): T-cell 활성화(ELISPOT 등 기능성 측정)으로 최종 목표를 직접 최적화
출처: IEDB, MHC BN, NCI, PRIME-2.0, 총 12,782 샘플.
✅ 4) 손실 함수(세 가지를 함께 씀)
Task-specific loss:
바이너리 과제(MHC, TCR, Hu-CLS, 미세조정)는 BCE(식 (4))
서열 마스킹 과제(EL, Hu-MLM)는 토큰 단위 CE(식 (5))
마스킹 80%·변이 10% 규칙
Orthogonal loss: 공유 인코더 임베딩 𝑒integrative 과
태스크 인코더 임베딩 𝑒task의 내적 절대값을 최소화해 표현 분리 유도(식 (6))
Adversarial loss: 소스 판별기는 CE로 출처를 맞히도록, 공유 인코더는 gradient reversal로 출처를 혼란 주도록 학습(식 (7))
도메인 불변 표현을 강화.
5) Fig. 1(스키마) 읽는 법
(a–f) 사전학습: EL-I/II, MHC-I/II, TCR, Hu-CLS/MLM을 동시에 학습
세 가지 학습 신호가 함께 흐름: (i) 태스크별 로스, (ii) 직교 제약(연녹색 라인), (iii) 소스 판별기·적대적 로스(진녹 라인)
(g–h) 미세조정: 공유 인코더를 T-cell 활성화(Task-IM)로 작게 재학습하고 전용 IM 디코더로 최종 면역원성 예측
이 전략은 일반화와 핵심 면역학 특징 학습을 동시에 확보
6) 추론 시 입력 유연성
실제 사용 시 펩타이드+MHC만으로 점수를 낼 수 있고, 필요하면 TCR 서열이나 species 레이블을 선택적으로 추가할 수 있음
요약 한 줄: 공유 인코더로 ‘도메인 불변’ 핵심 신호를 만들고, 도메인별 인코더·디코더로 보완 정보를 붙여, 적대적 학습+직교 제약으로 누수를 막으면서
①여러 전(前)단계 신호를 넓게 배우고
②최종 활성화로 좁혀서 다듬는 2단계 전략
✅ Biological Motivation and Justification for Multi-domain Pretraining
왜 다중-도메인 사전학습이 필요한가
1) 무엇을 분석했나
사전학습에 쓰인 데이터(EL, MHC, TCR)와 미세조정 데이터(IM) 사이에서 완전히 같은 9-mer가 들어있는 펩타이드를 찾아, 각 데이터의 라벨(예: “결합함/안 함”, “면역원성 있음/없음”)이 서로 얼마나 맞는지 비교(그림 2a)
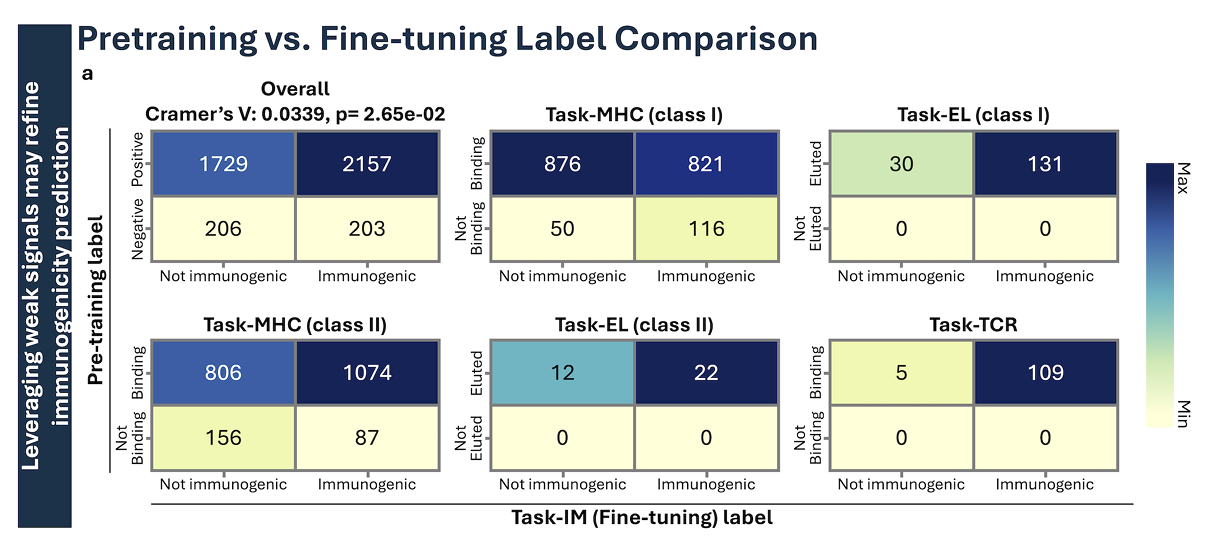
이유는 아미노산 1개 치환만으로도 면역원성이 크게 바뀔 수 있기 때문(그 사례가 Fig. 6에 제시됨)
2) 핵심 통찰 두 가지
라벨 인버전(label inversion)이 자주 보임
MHC 결합은 강한데 실제 T세포 활성(사이토카인 분비)은 유도되지 않는 펩타이드들이 많다
→ 즉, 한 단계(예: MHC 결합)만 보고는 면역원성을 제대로 못 맞춘다는 뜻
(면역원성 연쇄의 한 단계만 배운 모델의 근본적 한계)
이건 단순한 “데이터 누수” 문제가 아니라, 중간 단계 신호와 최종 면역반응 사이 관계가 비선형·복잡하다는 의미
중간 태스크 라벨과 최종 라벨의 상관은 약하지만 유의
전체적으로 Cramer’s V = 0.0339, p = 0.0265.
→ 중간 단계 데이터에도 유용한 힌트가 조금씩은 들어있다는 뜻
즉, 개별로는 약한 신호라도 잘 통합하면 예측에 분명 도움이 됨
이 점이 다중-도메인 프레임워크의 필요성을 뒷받침
결론(왜 다중-도메인인가)
한 가지 데이터(예: MHC 결합)만 학습하면 라벨 인버전 때문에 한계가 존재
그렇다고 중간 데이터가 쓸모없지는 않음
약하지만 진짜 있는 신호를 여러 도메인(EL, MHC, TCR, selfness 등)에서 모아 같이 학습하면, 최종 면역원성(IM)을 더 견고하고 정확하게 예측할 수 있음
→ 이게 다중-도메인 사전학습을 쓰는 이유
✅ Preventing Data Leakage: Curation and Splitting Strateg
왜 신경 쓰나
다중 도메인(EL/MHC/TCR/IM) 데이터를 합쳐 쓰면 데이터 누수(leakage) 위험이 커짐
그래서 저자들은 엄격한 큐레이션·분할 전략을 세워 리키지-프리 평가
(1) 누수 위험을 먼저 구조적으로 진단
IM(T-cell activation) 라벨은 면역반응의 최종 단계라서, 자연히 상위(activation) 데이터와 내용이 겹치고(intra-task leakage 위험), 하위 단계인 pMHC 결합·TCR-pMHC 인지 정보도 내포함(inter-task leakage 위험)
TCR-pMHC는 pMHC 결합 정보를 포함하지만 반대는 아님(pMHC 결합 데이터만으로는 TCR 인지/활성 정보를 알 수 없음)
이 비대칭을 고려해, 학습↔벤치마크 사이의 모든 누수 경로를 점검하고 intra/inter-task 누수 모두 제거
(2) 공통 원칙(가장 중요한 규칙)
아예 동일 펩타이드뿐 아니라, 9-mer 조각 기준으로 ‘두 자리 미스매치 미만’의 고유사 유사 서열도 전부 금지(세트 간 공유 금지)
(3) 데이터셋별 필터링(큐레이션)
pMHC 결합·EL(LC-MS): 비표준 아미노산·길이>20 제거, 짧은 건 20으로 패딩(모델 입력 길이 표준화)
HLA allele 주석 불충분하면 제외
TCR-pMHC: 비표준 아미노산/길이 기준으로 필터링, TCR CDR 서열 누락·품질 낮음(예: VDJdb 점수 <1)은 제외
Human vs Non-human(‘selfness’ 학습): 각 단백질을 겹치는 9-mer로 쪼개 사람/비사람에 모두 등장하는 9-mer 포함 펩타이드는 제외(종 특이 9-mer만 남김)
이 데이터도 IM처럼 누수에 보수적으로 대응
IM(미세조정용 T-cell 활성): 위 pMHC 필터와 동일 규칙 적용 후 사용
(4) 추가 초강수(훈련↔벤치마크 완전 분리)
훈련·벤치마크 모든 펩타이드 → 겹치는 9-mer들로 분해 → cd-hit로 클러스터링 → 벤치마크의 9-mer와 ‘두 자리 미스매치 미만’으로 겹치면 훈련 샘플에서 제외
(핵심 에피토프가 대개 9-mer에 들어있다는 가정)
이 기준은 NetMHCpan/BigMHC보다 더 엄격
(5) 최종 분할 방법
큐레이션 완료 뒤 train:val=9:1로 쪼개되, 분할 자체도 9-mer 수준 클러스터링으로 수행해 코어 서열이 양쪽에 겹치지 않도록 함(튜닝을 현실적이면서도 까다롭게 만들기 위해)
✅ TITANiAN-IM: T-cell Epitope Immunogenicity Prediction for Specific Peptide-MHC Pairs
특정 Peptide-MHC 쌍의 T-cell 면역원성 예측
✅ 왜 중요한가
네오안티젠(암) 발굴과 감염병 백신 설계에서 펩타이드-MHC 쌍의 면역원성을 아는 게 핵심
ELISPOT 같은 실험이 유용하지만 대량 처리에 비싸고 느림
→ 계산 예측 모델이 후보 수를 줄이는 데 큰 도움
✅ 모델 구성(무엇을 입력·출력하나)
TITANiAN-IM = 공유 인코더(Integrative encoder) + Task-IM 디코더 결합
입력은 펩타이드 서열 + MHC pseudo-sequence(펩타이드 접촉 잔기만 추린 대리 서열)이고, 출력은 면역원성 점수
같은 구조이되 사전학습 없이 미세조정 데이터만으로 훈련한 titanian-im을 비교용 베이스라인으로 둠
✅ 평가지표(4가지)
Mean PPVn(상위 n개 예측의 평균 정밀도), Precision, PR-AUC, AUROC
비교군에는 BigMHC, NetMHCpan-4.1, PRIME-2.0 등 SOTA 모델들이 포함
✅ 벤치마크 데이터(2세트)
Neoantigen discovery set: 833개(양성 178, 음성 655)
Infectious-disease vaccine discovery set: 2,093개(양성 1,488, 음성 605)
두 세트 모두 엄격한 누수 차단 하에 평가
✅ 누수 방지(평가 시)
훈련 데이터에서 벤치마크 펩타이드와 9-mer가 ‘두 자리 미스매치 미만’으로 겹치면 전부 제외
이렇게 해서 암기 효과가 아니라 진짜 일반화를 보게 함
또한 부트스트랩 1,000회로 95% CI를 추정하고, 순위 인접 모델 및 베이스라인과 쌍대 비교(Wilcoxon, Bonferroni 보정)를 수행
HLA 유형·펩타이드 길이 하위군별 분석도 수행
✅ 결과 요약
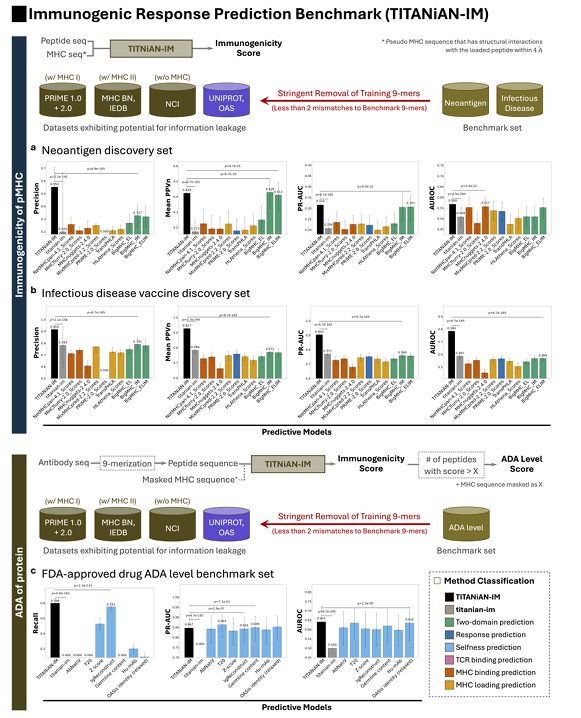
(A) 네오안티젠 벤치마크(Fig.4a)
사전학습 없는 titanian-im 대비: 모든 지표에서 유의하게 우수(p<0.001) → 사전학습의 효과가 큼
BigMHC-IM과 비교하면 Mean PPVn은 유사(0.424 vs 0.439)지만, Precision은 TITANiAN-IM이 훨씬 높음 등 지표 전반에서 우
(B) 감염병 백신 벤치마크(Fig.4b)
모든 지표에서 기존 방법을 상회
예를 들어 Precision 0.916, Mean PPVn 0.927로 BigMHC-IM(0.791, 0.771) 대비 크게 개선(둘 다 p<0.001)
PR-AUC 0.904 vs 0.760, AUROC 0.784 vs 0.569(BigMHC-IM/EL 대비)도 통계적·실용적으로 우수
오차막대(95% CI)가 타 모델보다 좁거나 비슷
→ 성능의 안정성도 높음
베이스라인(titanian-im) 대비 오차막대가 특히 작음
✅ 왜 이렇게 격차가 더 크나?
사전학습에 ‘human–nonhuman 구분’ 도메인이 포함돼 있어 병원체 유래 서열(비-self)을 면역원성으로 잘 식별하도록 일반화되었기 때문으로 해석
->
감염병 벤치마크의 “양성” 사례들은 대부분 병원체(=비-self)에서 온 펩타이드들
TITANiAN은 사전학습 단계에서 사람 단백질 vs 비-사람 단백질을 가르는 과제(Hu-CLS/MLM)를 배워서, 서열만 보고도 ‘self ↔ non-self’ 특징을 잘 잡아내도록 인코더가 길들여져 있음
그래서 비-self인 병원체 펩타이드를 만나면 “이건 자기 아닌데?
→ 면역원성일 가능성 높다”는 방향으로 일반화가 특히 잘 된다
→ 감염병 세트에서 성능 격차가 더 커진다는 해석
반대로 네오안티젠(암)은 사람 단백질에 생긴 미세한 변이(=거의 self)
-> 단순한 self/非self 구분만으로는 충분치 않음
self에 매우 가까운 것들 중에서 ‘조금 다른’ 것을 가려내야 하니, 사전학습 효과가 직접적으로 맞아떨어지는 정도가 상대적으로 작다
→ 감염병보다 격차가 덜 커 보임
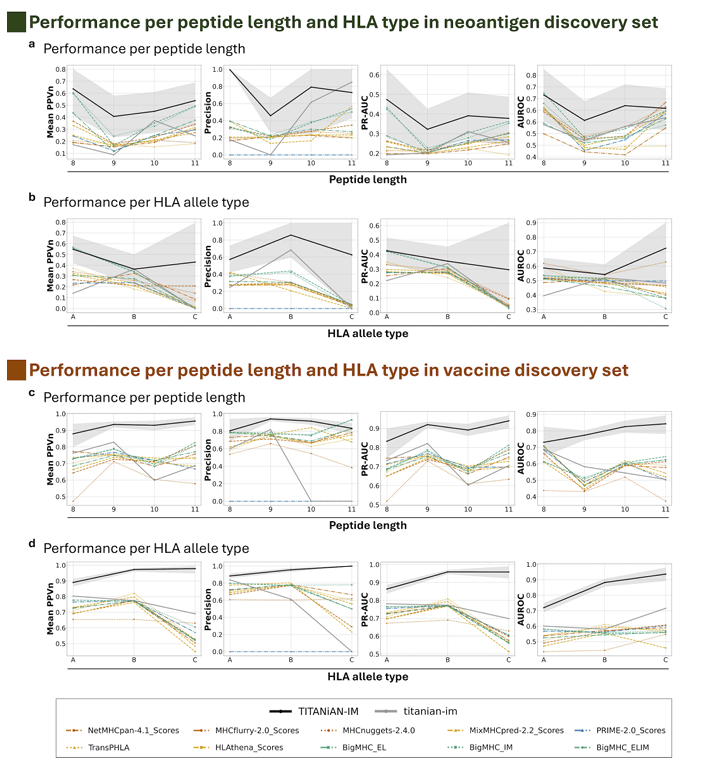
(C) 길이·HLA 유형별 강건성(Fig.5)
펩타이드 길이: 네오안티젠(Fig.5a), 감염병(Fig.5c) 모두에서 8–11mer 전 구간에서 지속적으로 강함
다른 모델들이 길이 제한(예: NetMHCpan 8–14aa, PRIME-2.0 8–11aa)이나 길이 증가에 따른 성능 저하를 보이는 반면, TITANiAN-IM은 최대 20aa까지 대응하며 사전학습에서 다양한 길이를 본 효과로 해석
HLA allele type: 두 벤치마크 모두에서 A/B/C 주요 유형 전반에 걸쳐 안정적·상대 우위(Fig.5b,d)
다만 네오안티젠의 HLA-C는 표본 양성 수가 매우 적어(Table D2), 일반화에 불리할 수 있음
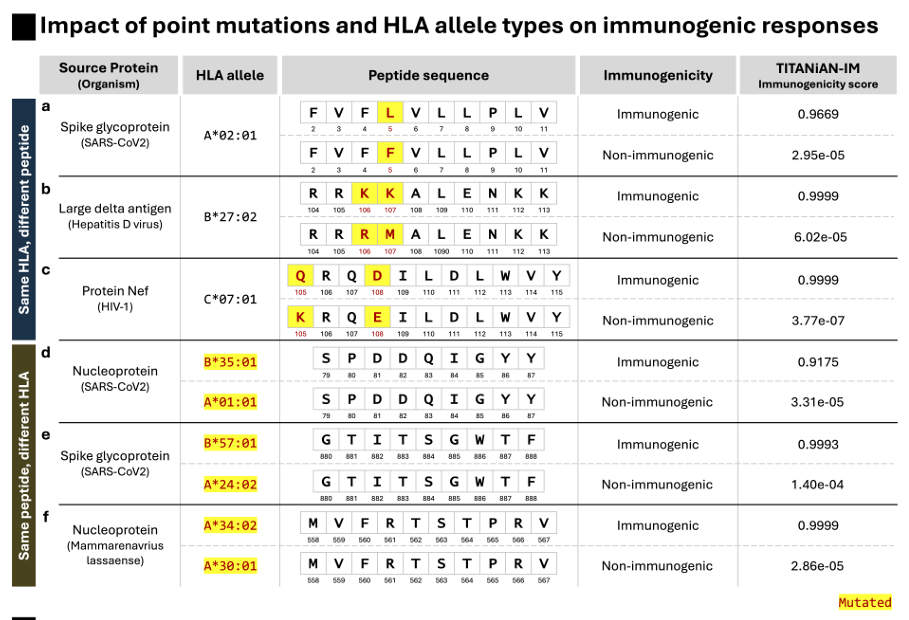
(D) 사례 연구(Fig.6)
점 돌연변이 1개로도 면역원성 점수 급변(예: SARS-CoV-2 Spike의 0.9669 vs 2.95e-05), 동일 펩타이드라도 HLA allele이 다르면 점수가 크게 달라짐(HLA-B35:01 vs HLA-A01:01).
→ 미세 변화/컨텍스트 차이를 정밀히 구분
(a–f) 점 변이 & HLA allele 차이가 면역원성을 어떻게 바꾸는지,
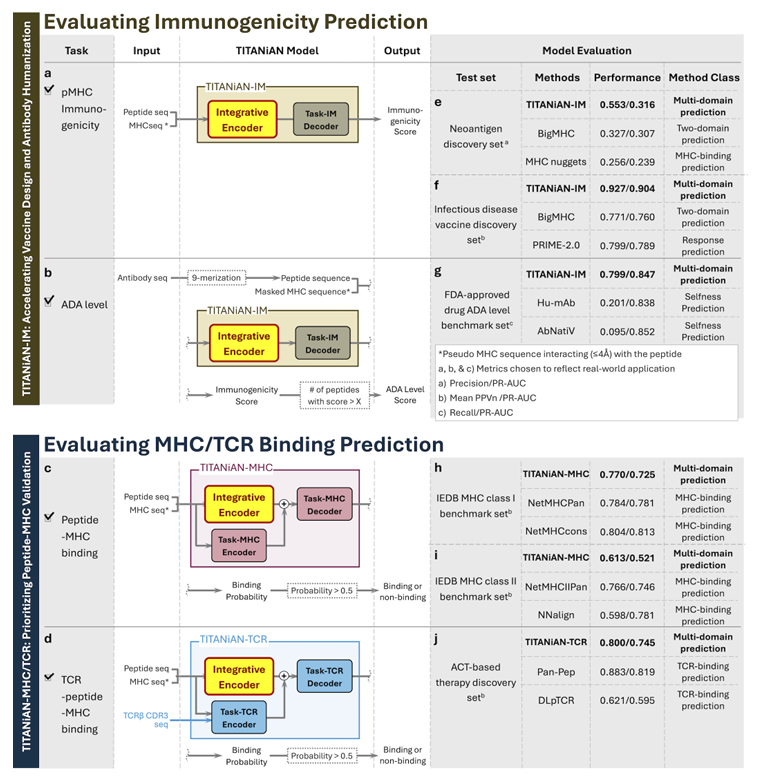
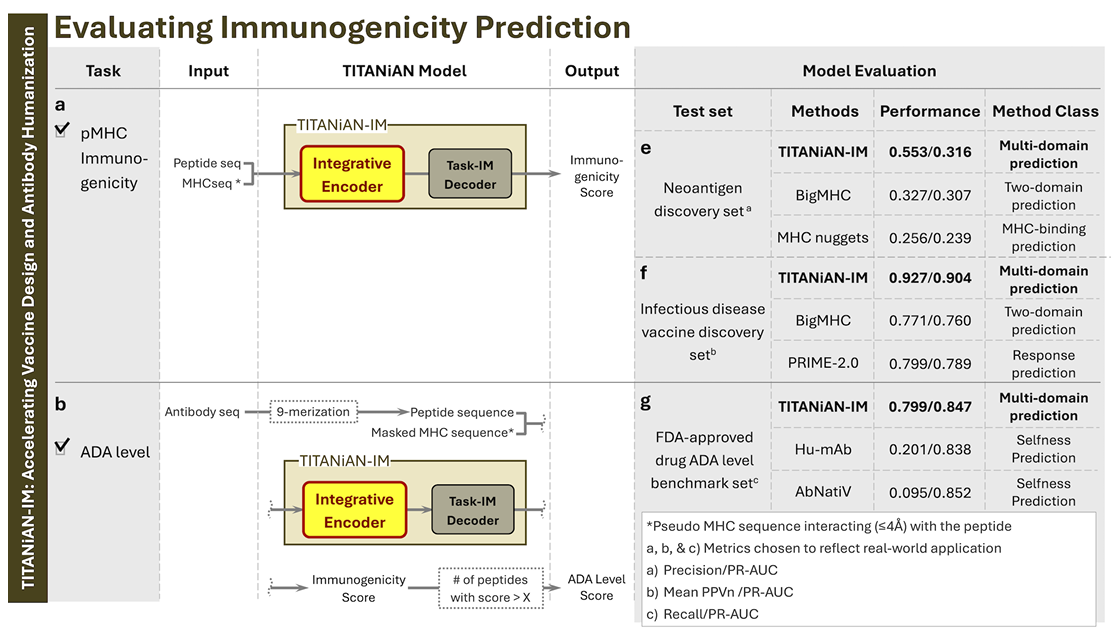
Fig.3 (구성 & 요약 성능)
(a) TITANiAN-IM: 공유 인코더 + Task-IM 디코더, 면역원성 점수 출력
(b) 항체 ADA 예측에도 같은 경로(Task-IM 디코더) 사용
(c, d) pMHC 결합(TITANiAN-MHC), TCR-pMHC 결합(TITANiAN-TCR)은 공유 인코더 임베딩과 태스크 인코더 임베딩을 연결해 태스크 디코더로 예측
(e–g, h–j) 각 태스크의 벤치마크별 성능 요약 패널 포함
요약
TITANiAN-IM은 공유 인코더+Task-IM 디코더로 펩타이드+MHC pseudo-sequence에서 면역원성을 직접 점수화함
두 개의 독립 벤치마크에서 누수 없이 평가했을 때, 정확도(Precision·PPVn)·랭킹(PR-AUC)·구분력(AUROC) 모두에서 기존 방법을 앞섰고, 길이·HLA 유형별로도 강건
특히 감염병 세트에서의 큰 격차는 human–nonhuman(자기/비자기) 사전학습 덕에 외래 항원 인지에 강해진 효과로 해석됨
✅ TITANiAN-IM: Application to Anti-Drug Antibody Level Prediction for Therapeutic Antibodies
1) 무엇을 어떻게 예측하나(방법)
목표: 항체의 ADA(anti-drug antibody) 생성 수준을 예측
방식: 항체 아미노산 서열을 9-mer로 슬라이딩해 잘라서, 각 조각의 면역원성 점수를 계산하고 0.5 초과인 조각들을 “면역원성 조각”으로 간주
이때 MHC pseudo-sequence(알렐 의사서열) 입력이 아예 필요 없다는 것이 핵심
왜 이렇게 하냐: 항체 서열 안의 잠재적 면역원성 구간을 빠짐없이 잡아내는 것이 목적이므로, Recall(재현율)을 주평가지표로 둔다(모든 위험 부위를 놓치지 않는 게 더 중요)
2) 어떤 데이터·평가 기준을 썼나
벤치마크: FDA 승인 항체 216종(양성 169 / 음성 47)
비교군: titanian-im(사전학습 없는 동일 구조 베이스라인) + 기존 selfness 기반 방법들(AbNatiV, T20, Z-score, IgReconstruct, AbLSTM, Germline content, Hu-mAb, MG Score, OASis).
항체를 ‘면역원성’으로 판정하는 규칙: 해당 항체의 9-mer들 중 20% 초과가 면역원성 점수 > 0.5이면 그 항체는 면역원성(양성)으로 간주(Prihoda et al. 기준 따름)
누수 방지: 평가 전에 벤치마크 9-mer와 ‘두 자리 미스매치 미만’으로 겹치는 모든 훈련용 펩타이드는 제외
통계 검정: 부트스트랩 1,000회로 95% CI 추정 + 쌍대 Wilcoxon(Bonferroni 보정)
필요 시 직하·직상(한 단계 아래/위 순위 모델), 그리고 베이스라인(titanian-im)과의 쌍대 비교로 유의성 확인
3) 결과(Figure 3, 4)
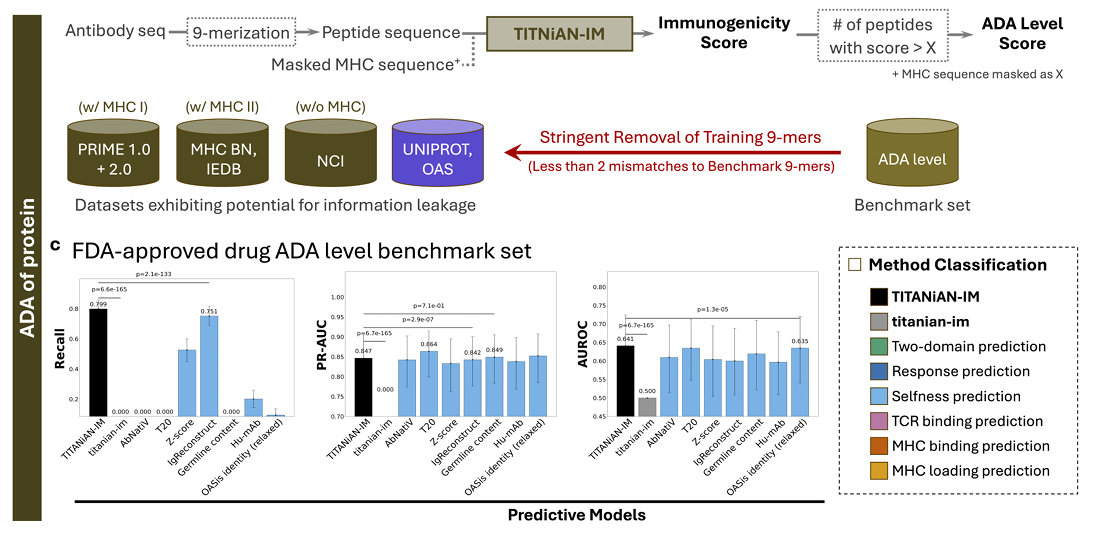
입력: 항체 서열 → 9-merization → (MHC 서열은 마스킹/불사용)
모델: Integrative encoder + Task-IM decoder
출력: (i) 각 9-mer의 면역원성 점수, (ii) 임계치 초과 9-mer 개수를 집계해 ADA level score로 활용(벤치마크에서 Recall 등으로 평가)
TITANiAN-IM은 모든 지표에서 다른 방법들을 ‘일관되게·유의하게’ 앞섬(교정 p<0.001)
사전학습 없는 베이스라인(titanian-im)은 면역원성 항체를 하나도 못 잡음
→ Recall=0, PR-AUC=0, AUROC=0.5(랜덤). 사전학습의 필요성이 명확
TITANiAN-IM 성능 수치:
Recall 0.799, 다음 순위 IgReconstruct 0.751보다 유의하게 높음(p<0.001)
PR-AUC 0.847(T20의 0.864와 비슷)
AUROC 0.641(OASis identity 0.635보다 유의하게 높음)
오차막대(95% CI)는 다른 모델과 비슷하거나 더 좁아 → 성능이 안정적
4) 사례(Fig. 6g–j) — 디이뮤나이즈(DAA) 검증

실험적으로 디이뮤나이즈된 아미노산(DAA)가 알려진 Etaracizumab, Bevacizumab, Pembrolizumab, Omalizumab에서, TITANiAN-IM이 DAA를 70–100% 회수
예측은 면역원성 점수>0.5인 9-mer 기반이며, 디이뮤나이즈 전/후 점수 분포가 뚜렷이 분리되어 예측의 해석력/타당성을 뒷받침
5) 왜 의미 있나(설계의 장점)
이 경쟁력 있는 성능이 MHC pseudo-sequence 없이 달성됨
→ 다중 도메인 사전학습(특히 MHC 비의존적 Task-Hu, 즉 human–nonhuman selfness 학습)으로 항체 서열만으로도 면역신호를 포착하도록 인코더가 일반화되었기 때문
치료용 항체 면역원성↓ 설계에 직접적인 실무 가치가 있음을 시사
한 줄 정리
TITANiAN-IM은 항체 서열만으로 면역원성 구간을 조각 단위(9-mer)로 찾아 ADA 위험을 잘 예측함
사전학습 없는 동일 구조는 완전히 실패(Recall=0)하지만, 다중-도메인 사전학습을 거친 TITANiAN-IM은 Recall 0.799, PR-AUC 0.847, AUROC 0.641 등 안정적·우수한 성능을 보였고, 실제 DAA 사례에서도 70–100% 회수해 설계적 해석력까지 입증
✅ TITANiAN-IM: Dataset Ablation Study
1) 무엇을 제거(ablated)했나 — 4가지 변형
사전학습 단계에서 특정 도메인 데이터+태스크를 하나씩 뺀 뒤, 그 태스크 전용 인코더/디코더도 모델에서 제거하고(공유 인코더는 유지) 같은 IM 미세조정을 다시 수행
만든 변형은 네 가지:
- TITANiAN-IM Hu: Task-Hu(사람/비사람 selfness 데이터) 제거
- TITANiAN-IM MHC I: MHC-I 결합·EL 데이터 제거
- TITANiAN-IM MHC II: MHC-II 결합·EL 데이터 제거
- TITANiAN-IM TCR: TCR–pMHC 결합 데이터 제거
2) 어떻게 평가했나 — 3개 벤치마크 × 5개 지표
세팅 3개:
(a) 네오안티젠(실험이 CD8+ 활성 → 주로 MHC-I 경로)
(b) 감염성 질환 백신(역시 CD8+ / MHC-I)
(c) FDA 승인 항체 ADA 레벨(환자 ADA 유도= B세포 활성 ← CD4+ 도움 → 주로 MHC-II 경로)
지표 5개: Mean PPVn, Precision, PR-AUC, AUROC, Recall
부트스트랩 1,000회로 95% CI, 쌍대 Wilcoxon + Bonferroni 보정으로 유의성 표시
(Fig.7)
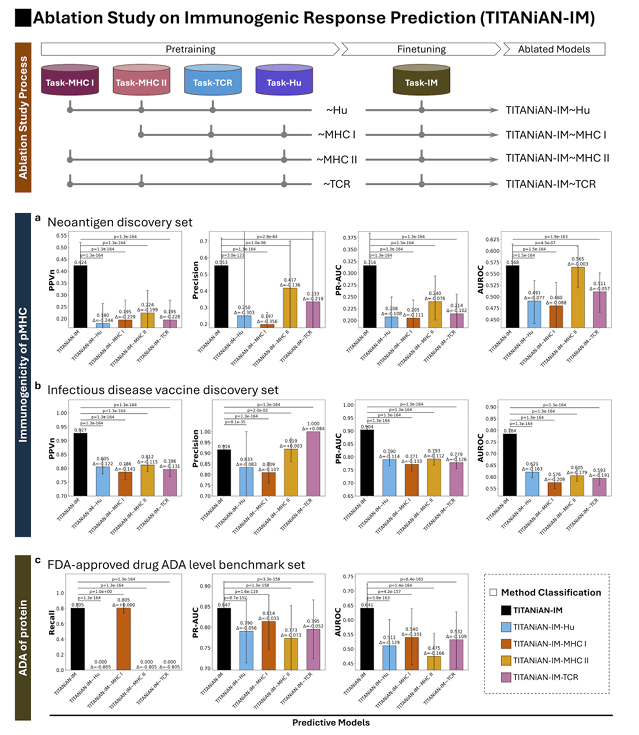
3) 무엇이 드러났나 — 핵심 결과(요약)
모든 도메인이 성능에 기여: 네 개 변형 모두 원본보다 유의하게 하락(모든 비교에서 p<0.001)
→ 각 도메인 신호가 정말 필요함
Task-Hu(사람/비사람) 제거 효과가 가장 광범위: 세 벤치마크 전반에서 하락이 큼
Self/Non-self 구분이라는 약하지만 넓은 신호가 일반화에 크게 기여
MHC-I 제거: CD8+ 관련 두 벤치마크((a),(b))에서 큰 성능 저하, CD4+ 중심 ADA 세트((c))에는 영향 제한적
MHC-II 제거: 반대 패턴—ADA 세트에서 뚜렷한 하락, (a),(b)에는 상대적으로 영향 적음
→ 모델이 클래스 I/II의 생물학적 차이를 제대로 익혔음을 시사
TCR 제거: 유의한 하락은 있으나 다른 도메인 제거 대비 폭은 작음
현 구조가 고변이 TCR CDR 정보를 충분히 표현하지 못하는 한계 때문일 수 있다고 해석
요약
Selfness(Hu) 신호가 가장 넓게 받쳐주고, MHC-I/II 신호는 각자 해당 경로(CD8+/CD4+)가 중심인 과제에서 결정적이며, TCR 신호는 보완적 역할을 한다
그래서 모든 도메인을 함께 사전학습하는 현재 설계가 최적이라는 결론
✅ TITANiAN-MHC: Predicting Peptide-MHC Binding
(1) 무엇을, 왜 하나
문제의식: 펩타이드–MHC 결합 예측은 면역원성 후보 발굴의 첫 관문이자(암 면역치료·백신 설계 등), Fig. 4a,b가 보여주듯 면역원성 직접 예측 모델도 아직 완벽하진 않음
그래서 보완 전략으로 “결합 친화도 기반 사전 선별(pre-filtering)”이 필요하다고 밝힘
(2) 왜 선별이 유용?
결합 예측으로 실험(다운스트림 면역원성 테스트) 탐색 공간을 크게 줄일 수 있음
이 맥락에서 정확히 ‘진짜 결합자(true binder)’를 잘 골라내는가가 중요하므로, Precision과 Mean PPVn(상위 n개 평균 정밀도)를 핵심 지표로 삼고, 이 둘을 올리면서 재현율이 희생되지 않도록 PR-AUC도 함께 본다고 명시
분류 전반 성능은 AUROC로 보조 평가
(3) 모델 구성(그림 3c)
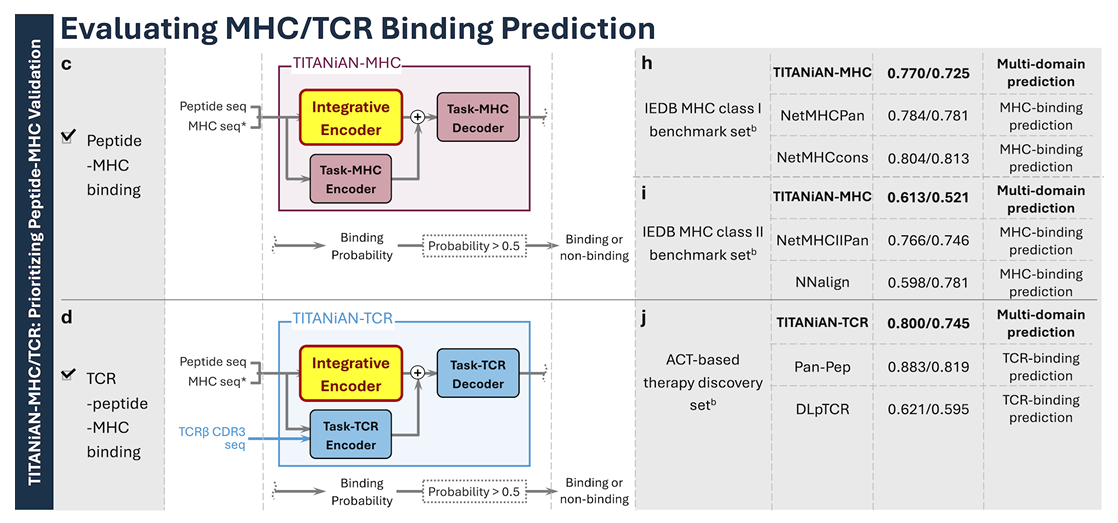
TITANiAN-MHC는 공유 인코더(Integrative encoder) + Task-MHC 전용 인코더/디코더 조합을 사용
중요: 여기서는 태스크별 미세조정 없이, 오로지 다중-도메인 사전학습으로 얻은 파라미터만으로 동작하도록 평가함
(4) 평가 셋·누수 차단·통계 절차
벤치마크: IEDB의 신규 수집분으로 리키지 위험을 피함
Class I: 총 624(양성 344, 음성 280) — 2020-12-25 이후 수집 자료.
Class II: 총 723(양성 242, 음성 481) — 2021-02-19 이후 수집 자료.
리키지 방지: 벤치마크의 어떤 펩타이드와도 9-mer 기준 미스매치 < 2로 겹치는 훈련 시퀀스는 전부 제외(일반화 검증을 위한 엄격 기준)
또한 다양한 HLA 분포가 되게 큐레이션했고, Class I 벤치마크에 HLA-E를 포함(학습에서 저대표(under-represented) 된 알렐로 일반화 능력을 점검하려는 의도)
통계: 부트스트랩 1,000회로 95% CI, 쌍대 Wilcoxon + Bonferroni 보정
특히 성능 바로 위·아래 모델과의 쌍대 비교로 유의성을 판정
(5) 결과 (Fig. 8a–b)
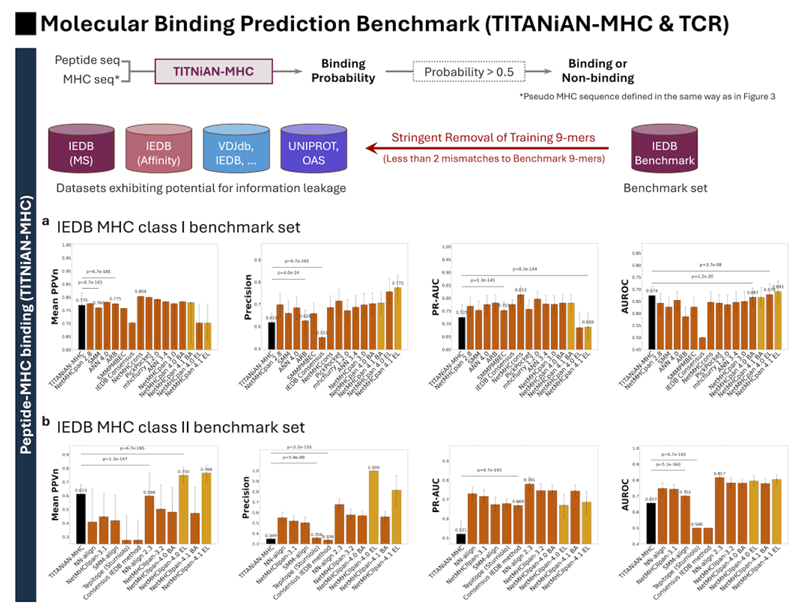
Class I 결합 예측 (Fig. 8a)
비교군: NetMHCpan 2.8/3.0/4.x, SMM, ANN4.0/3.4, ARB, SMMPMBEC, IEDB Consensus, NetMHCcons, mhcflurry 1.2.0 등
TITANiAN-MHC I 성능:
Mean PPVn = 0.770 (IEDB Consensus 0.804보단 낮지만, 여러 NetMHCpan 버전과 비슷, 더 낮은 모델들보단 유의하게 우수, p < 0.001)
Precision = 0.619 (NetMHCpan 4.0 EL의 0.775보다 낮음)
PR-AUC = 0.725 (NetMHCcons 0.813과 비교 가능한 수준, NetMHCpan 4.0 EL보다는 높음, p < 0.001)
AUROC = 0.674, NetMHCpan 4.1 EL 0.691과 유사, NetMHCpan 4.0 등 이전 세대 모델보단 높음
주목할 점: 태스크별 미세조정 없이도(=사전학습 지식만으로) 위 성능을 달성
Class II 결합 예측 (Fig. 8b)
비교군: NN-align 2.3, NetMHCIIpan-3.1/4.0 EL/4.1 EL, SMM-align, Tepitope, IEDB Consensus.
TITANiAN-MHC II 성능:
Mean PPVn = 0.613 (NN-align 2.3의 0.598보다 유의하게 높음, p < 0.001), 전체 순위로는 3위(NetMHCIIpan-4.0 EL 0.750, 4.1 EL 0.766 다음)
Precision = 0.349, PR-AUC = 0.521(각각 NetMHCIIpan-4.0 EL의 1.000, NN-align 2.3의 0.781보단 낮음)
그래도 IEDB Consensus(precision 0.338) 등 몇몇 방법보단 우수(p < 0.001)
AUROC = 0.657으로 NetMHCIIpan-4.0 EL 0.817보다 낮지만 Tepitope 0.500보단 유의하게 높음(p < 0.001)
해석: Class II에서 격차가 상대적으로 큰 이유는 펩타이드 길이·결합 양식 변동성이 크기 때문이라고 설명
한 줄 정리
TITANiAN-MHC는 사전학습만으로 Class I에선 상당히 경쟁력, Class II에선 전반적으로 준수한 성능을 보이며, 결합 기반 사전 선별을 위한 Precision/PPVn 중심의 지표에서 강점이 확인(누수 철저 차단, 통계 유의성 검정 포함)
✅ TITANiAN-TCR: Predicting TCR-Peptide-MHC Binding
1) 문제의식
TCR–pMHC 결합 예측은 ACT(Adoptive Cell Transfer) TCR 설계나 감염 질환 코호트 분석에 직접 쓰일 수 있어 실용성이 큼
이런 응용에선 신뢰도 높은 결합(고확신 양성)을 찾는 게 핵심이라 Precision과 Mean PPVn을 주 지표로 삼고, 분류 성능 전반 평가를 위해 PR-AUC, AUROC도 함께 봄
이 데이터셋은 양·음성 비율이 비교적 균형적이라 AUROC도 중요 지표로 강조한다.
2) 모델 구성
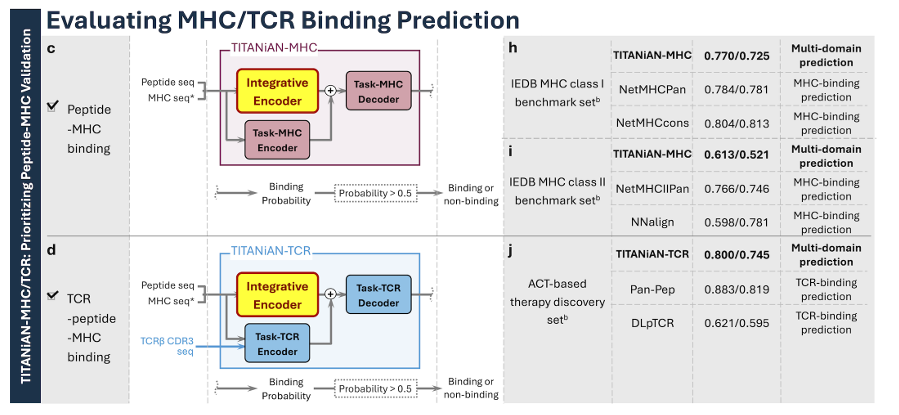
TITANiAN-TCR = 공유 integrative encoder + Task-TCR 전용 인코더/디코더(Fig. 3d)
중요: 태스크별 미세조정 없이, 사전학습으로 얻은 파라미터만 사용해 예측을 수행
3) 벤치마크와 비교 대상
Panpep zero-shot 테스트: 총 786 (양성 393 / 음성 393)
ACT test: 총 26 (양성 13 / 음성 13)
ACT 기반 면역치료 개발과 가장 직접적으로 연결되어 중요도를 높여 평가
비교군: Pan-pep, DLpTCR, pMTnet, ERGO2
4) 통계 절차
부트스트랩 1,000회로 95% CI 추정 + 쌍대 Wilcoxon(Bonferroni 보정)으로 근접 성능 모델들과의 유의성 검정
5) 결과
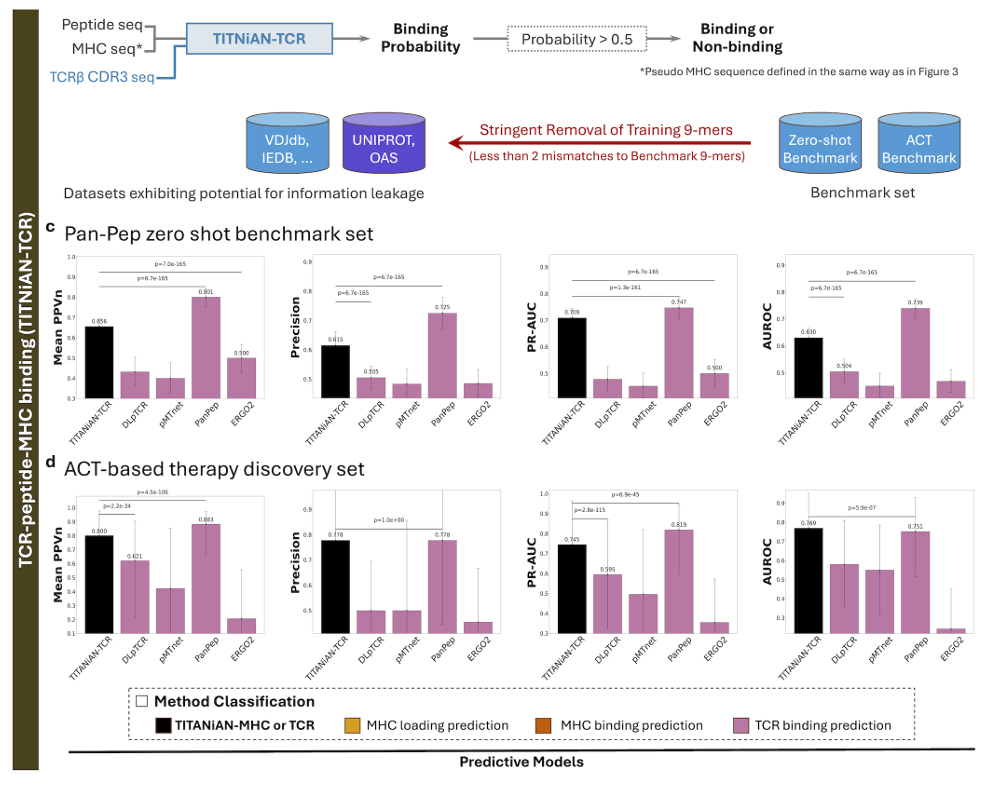
A) Zero-shot 벤치마크 (Fig. 8c)
Mean PPVn: 0.656 — Pan-pep 0.801에 이어 2위, ERGO2 0.500보다 유의하게 높음 (p<0.001)
Precision: 0.650 — Pan-pep 0.717보단 낮지만 DLpTCR 0.505보다 유의하게 높음 (p<0.001)
PR-AUC: 0.709 — Pan-pep 0.747과 근접, ERGO2 0.500보다 유의하게 높음 (p<0.001)
AUROC: 0.630 — Pan-pep 0.739을 제외한 모든 모델보다 높음(예: DLpTCR 0.504; p<0.001)
해석: Zero-shot에서는 Pan-pep의 메타러닝 최적화 때문에 Pan-pep이 앞서지만, TITANiAN-TCR도 전반적으로 강력
B) ACT-based therapy discovery 벤치마크 (Fig. 8d)
Mean PPVn: 0.800 — Pan-pep 0.883에 근접, DLpTCR 0.621보다 유의하게 높음 (p<0.001)
Precision: 0.778 — Pan-pep와 동일, 다른 모든 모델보다 유의하게 높음 (p<0.001)
PR-AUC: 0.745 — Pan-pep 0.819과 비교 가능, DLpTCR 0.595보다 유의하게 높음 (p<0.001)
AUROC: 0.769 — 모든 모델 중 최고, Pan-pep 0.751보다 유의하게 큼 (p<0.001)
→ binder/비-binder 구분력에서 통계적으로 유의한 우위
다만 다른 지표들에선 Pan-pep와 대체로 비슷한 수준
6) 설계적 의미(저자 해석)
이 성과는 특히 주목할 만한데, Task-TCR 모듈을 ‘보조(auxiliary)’로 설계했고, 핵심 인코더는 pMHC 입력만 받으며 TCR 특화 처리는 최소화했음에도 TCR 과제에서 합리적인(경쟁력 있는) 성능을 달성했기 때문
즉, 가벼운 설계로도 충분한 수준의 예측력을 확보함을 보임
요약
TITANiAN-TCR은 사전학습만으로 zero-shot에서도 상위권, ACT 실제 데이터에선 정밀도·랭킹·구분력 모두 SOTA에 근접 또는 우위(특히 AUROC 최고)를 보임
TCR 특화 파라미터가 적은 ‘경량’ 설계임에도 실전적 성능을 확인
✅ Discussion
1) 무엇을 제시했나
이 연구는 TITANiAN이라는, 다양한 생물학적 정보를 통합해 T-cell 에피토프 면역원성을 예측하는 포괄적 딥러닝 프레임워크를 소개
미세조정 모듈(TITANiAN-IM) 기준으로 주요 예측 과제에서 성능을 입증:
특정 peptide-MHC 쌍의 T-cell 활성 예측에서 SOTA 달성, 기존 방법을 자주 능가했고,
MHC 입력 없이도 치료용 항체의 면역원성(ADA 유발 가능성)을 상위권 성능으로 평가
또한 다중 도메인 사전학습 덕분에 분자 결합 같은 추가 면역 관련 예측 작업도 지원
이는 면역원성의 여러 측면에서 보이는 광범위한 효과가 사전학습 전략의 힘을 보여줌
2) 왜 잘 되나(설계 포인트와 견고성)
프레임워크는 모듈형 멀티태스크 구조에 적대적 도메인 적응을 접목해, 여러 데이터셋을 큐레이션하여 통합
리키지 방지형 데이터 큐레이션과 체계적 소거(ablations)로 각 사전학습 태스크의 기여를 검증
3) 한계와 향후 과제(개선 방향)
데이터 종류 확장:
TAP(수송체 연관 항원 처리) 데이터나 peptide-protein 도킹 정보를 포함하면 생물학적 이해를 풍부화할 수 있으나, 현 시점 데이터 가용성이 제한적
최적화 한계:
성능은 강하지만, 하이퍼파라미터 최적화로 추가 개선 여지가 있음
다만 사전학습 계산비용(예: RTX A6000에서 10 GPU-days)이 커서, 광범위한 최적화·탐색(태스크 조합 등)을 현재로선 제약
4) 기대 효과(벤치마크를 넘어)
TITANiAN은 결합→활성→selfness 신호를 통합하는 정확·포괄적 면역원성 예측 능력으로 생의학 발견을 가속할 잠재력이 있음
구체적으로,
단백질 치료제의 디이뮤나이제이션,
백신의 합리적 설계,
새로운 암 네오안티젠의 발굴을 촉진하여 더 안전하고 효과적인 치료에 기여할 수 있다고 전망
요약
TITANiAN은 다중 도메인 사전학습 + 적대적 도메인 적응을 통해 면역원성 예측 전반(SOTA 수준의 T-cell 활성, MHC 없이 항체 ADA 예측, 결합 과제 지원)을 견고하게 수행하며, TAP/도킹 데이터 추가와 더 넓은 최적화가 향후 성능 상향의 열쇠로 제시
https://galaxy.seoklab.org/design/t-scape/
✅ Method
✅ Dataset and task
공통 원칙
모든 데이터는 공개 소스에서 수집했고(표 D1 참조), 벤치마크와의 중복(데이터 리키지) 제거를 위해 사전에 9-mer 유사성 기반 엄격 필터링(§2.3, §4.3)을 적용함
학습/검증 어느 쪽에도 벤치마크와 겹치는 서열이 없도록 처리
✅ Cramer’s V analysis of label concordance
생물학의 중간 단계(예: p–MHC 결합, TCR–pMHC 상호작용)의 라벨과 면역원성(사이토카인 분비) 라벨 사이의 통계적 연관성을 정량화
겹치는 펩타이드를 바탕으로 연관표(contingency table)를 만들고 Cramer’s V를 계산
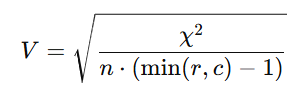
결과: 약하지만 유의한 연관을 관찰
Cramer’s V = 0.0339, p = 0.0265
이는 상류 태스크의 약한 신호도 면역원성 예측에 도움이 될 수 있음을 시사
✅ Sequence identity filtering using CD-HIT(누수 방지 필터링)
목적/가정: 학습·벤치마크 간 데이터 리키지 방지. 면역원성 신호가 핵심 9-mer에 국한되는 경우가 많다는 가정하에 9-mer 단위 유사도로 필터링
✅ Architecture of TITANiAN (모델 구조)
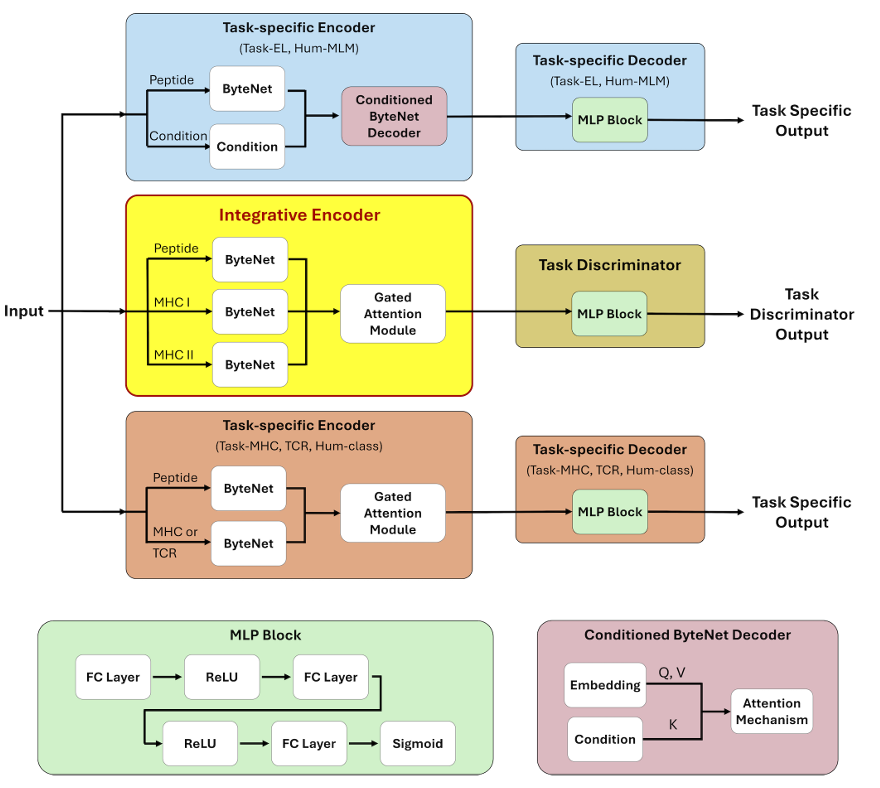
✅ 왜 CNN?
대부분의 단백질 LM은 Transformer/LSTM을 쓰지만, 이 과제는 긴 단백질을 9-mer로 쪼개 쓰므로 글로벌 어텐션이 꼭 필요하지 않음
로컬 패턴에 강한 CNN(커널 크기 3)을 채택했고, 기존 연구에서도 CNN이 경쟁력을 보였다고 인용
✅ 핵심 아키텍처: ByteNet
ByteNet 인코더–디코더를 공유(Integrative) 인코더와 태스크별 인코더의 코어로 사용
단, 조건부 마스킹 예측(EL, Hu-MLM)엔 조건 입력을 임베딩에 주입하기 위해 어텐션 모듈을 추가한 “conditioned ByteNet Decoder”를 사용, 그 외 태스크는 기본 디코더 사용
✅ 입력 임베딩 & 게이트드 어텐션 결합
Integrative encoder는 세 개의 ByteNet으로 펩타이드, MHC-I pseudo-seq, MHC-II pseudo-seq를 각각 임베딩
이 임베딩들을 게이트드 어텐션(gated attention)으로 결합
가중합으로 최종 임베딩 𝑒final 을 산출
MHC가 필요 없는 태스크(예: Hu-CLS)는 펩타이드 임베딩만 사용
ByteNet이 만든 임베딩은 어텐션의 query/value, 조건(condition)은 key로 사용
✅ 태스크별 경로
여러 입력이 필요한 태스크에서도 게이트드 어텐션을 태스크별 인코더에 적용
이후 공유 인코더 임베딩 + 태스크별 인코더 임베딩을 연결(concatenate)
→ 태스크별 디코더(MLP 1개 층)로 분류/회귀 수행
✅ 적대적 학습(도메인 불변화)
Discriminator(간단한 MLP)가 공유 인코더 출력을 받아 데이터 출처(source)를 분류하도록 학습
학습 시 gradient reversal을 걸어 인코더가 출처 판별이 안 되도록(=도메인 불변 표현) 유도 → 강건성·일반화 향상
✅ Training scheme
멀티도메인 적대적 학습(multi-domain adversarial learning)을 적용
손실은 세 가지로 구성: 태스크별 손실, 직교(orthogonal) 손실, 적대적 손실
1) Task-specific loss (태스크별 손실)
이진 분류 태스크(MHC, TCR, Hu-CLS, 그리고 파인튜닝 단계)는 Binary Cross-Entropy
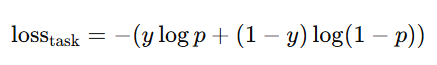
시퀀스 마스킹 예측 태스크(EL, Hu-MLM)는 토큰 단위 Cross-Entropy
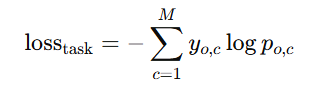
c=토큰(아미노산 코드 + stop/gap/mask/start 토큰까지),
𝑝𝑜,𝑐=각 토큰에 대한 모델 확률,
𝑦𝑜,𝑐=정답 토큰
Devlin et al. 방식을 따라 80% 마스킹, 10% 변이(mutated)를 적용
2) Orthogonal loss (직교 손실)
목적: 공유 인코더와 태스크별 인코더 표현을 분리(디스엔탱글)하기 위해 직교 제약 부여
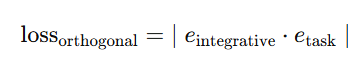
𝑒integrative=공유 인코더 임베딩, 𝑒task=태스크별 인코더 임베딩
내적이 0에 가까워질수록 서로 독립
3) Adversarial loss (적대적 손실)
목적: 공유 인코더가 데이터 출처(도메인) 정보를 못 담도록(=도메인 불변 표현) 유도
방법: 디스크리미네이터에는 (EL의 식과 유사한) Cross-Entropy를 적용하고, 공유 인코더에는 아래 적대적 손실을 걸어 gradient reversal로 학습
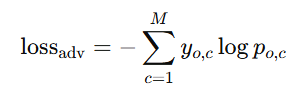
여기서
𝑐=태스크 인덱스,
𝑝𝑜,𝑐=각 클래스(데이터 소스) 확률,
𝑦𝑜,𝑐=정답 클래스 라벨
4) 최종 손실 결합
총 손실
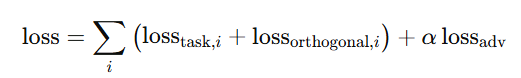
α=5 (값 1과 5를 시험 후 5로 설정)
더 큰 𝛼가 통합 인코더의 도메인 정렬(alignement)을 더 강하게 강제해 데이터 출처 간 표현을 잘 맞춘다는 점에 기인
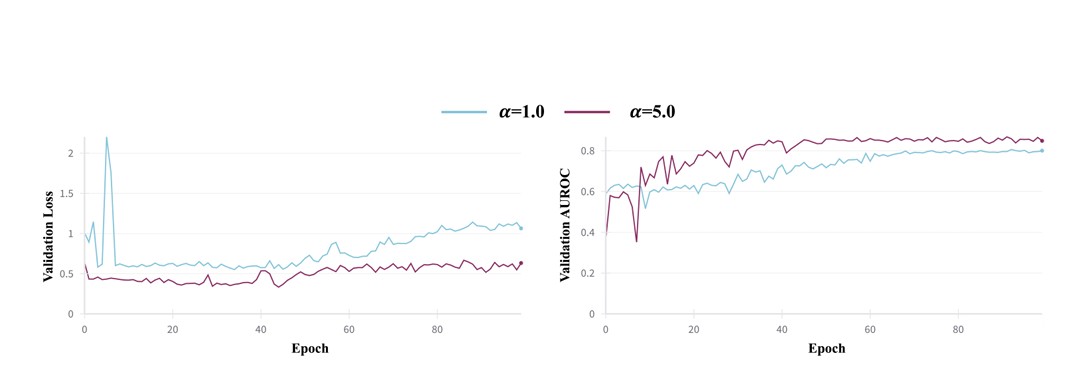
5) 구현/학습 설정(사전학습)
프레임워크: PyTorch. Adam 옵티마이저 + Cosine annealing 스케줄러.
하이퍼파라미터: LR 1e-5, 배치 40, gradient accumulation 4, 모델 차원 280, 6층, RTX A6000에서 총 10 GPU-days
6) 파인튜닝(미세조정)
손실: BCE(위 식(4)과 동일), 옵티마이저/스케줄러 동일 사용.
하이퍼파라미터: LR 5e-5, 배치 128, 100 epochs
✅ Benchmark method
✅ Benchmark label — 라벨/점수 표준화 규칙
모든 태스크를 분류 문제로 통일하기 위해, 연속 측정치/점수를 이진 라벨 또는 확률 유사(normalized) 스코어로 변환
ADA(anti-drug antibody) 평가셋
항체별 ADA 반응 인원 수 기반
0이 아닌 값 ⇒ 1(면역원성), 0.0 ⇒ 0(비면역원성)
다른 모델(주로 human-likeness 점수 출력)을 면역원성 스케일과 맞추기 위해 점수 변환을 적용
점수 변환 규칙(방향성 표준화)
사람스러움이 클수록 값↑(AbNatiV, Hu-mAb, Germline content 등): Transformed=1−𝑥
백분위 점수(예: T20):
Transformed=1−(𝑥/100)
Z-score류(Z-score, IgReconstruct, AbLSTM): 표준정규 CDF Φ 사용
Z-score/IgReconstruct: 1−Φ(x)
AbLSTM: Φ(x)
OASis identity(느슨→엄격): 1−𝑥
MHC 결합(IC50) 벤치마크 표준화
라벨링: IC50 ≤ 500 nM ⇒ 1(bind), > 500 nM ⇒ 0(non-bind) (IEDB 가이드에 따르는 일반 임계값)
모델 출력(IC50 예측) 정규화: Transformed=1−𝜎(𝑥−500)
→ 결합 강함(작은 IC50)일수록 1에 가깝게 매핑되어 분류기 기반 모델과 비교 가능
위 변환으로 모든 데이터·모델을 하나의 분류 프레임에서 일관되게 평가
✅ Embedding quality benchmark — 임베딩 품질 평가
대상: Integrative encoder가 만든 임베딩
이를 t-SNE(2D)로 투영한 뒤, SI와 AMI 두 지표로 군집 품질을 정량화
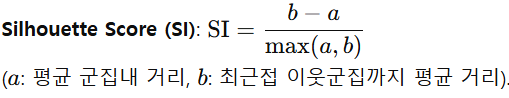

✅ Appendix D — 다양한 데이터셋에서의 t-SNE 임베딩
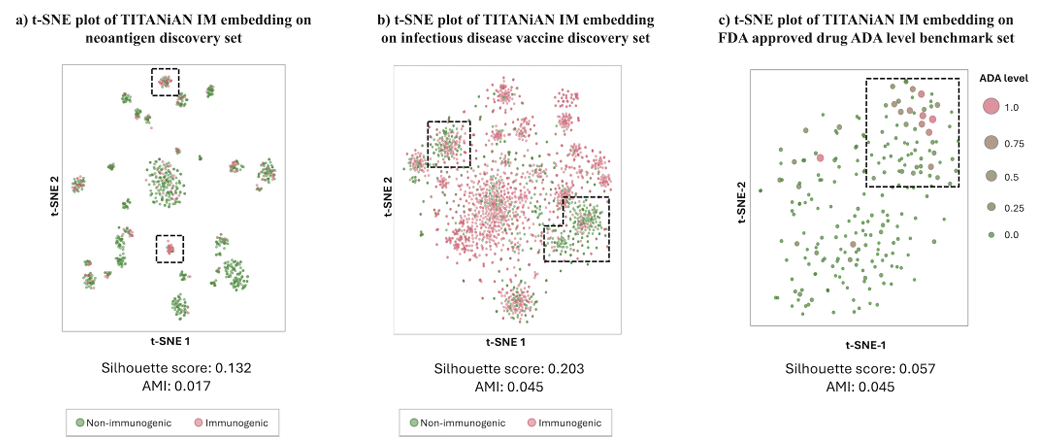
TITANiAN-IM의 성능이 디코더 분류기만의 결과가 아니라, 사전학습+파인튜닝으로 얻은 임베딩의 품질에도 좌우됨을 보이기 위해, 통합 인코더의 출력 텐서를 사용해 t-SNE 시각화를 제시(Fig. D5).
네오안티젠 세트(Fig. D5a):
면역원성 펩타이드가 서로 뭉치고, 비면역원성 점들은 그 영역에 최소한만 섞임
SI=0.132, AMI=0.017으로, 무작위 임베딩(SI=−1.0, AMI=0.0) 대비 군집 품질 우수
감염성 질환 백신 세트(Fig. D5b):
비면역원성 펩타이드가 뚜렷한 군집을 형성하고 면역원성 점이 거의 섞이지 않음
SI=0.203, AMI=0.045(무작위 대비 우수)
ADA 벤치마크(항체)(Fig. D5c):
항체 서열은 각 항체의 펩타이드 시퀀스에 대해 max-pooling으로 단백질 임베딩을 만들었고, 문제 유발 서열이 한 곳만 있어도 면역원성이 생길 수 있음을 반영
SI=0.057, AMI=0.045로 무작위 대비 우수, 면역원성 관련 구조를 포착한 의미 있는 임베딩을 생성함을 시사
결론: 위 분석은 임베딩 자체가 면역원성/비면역원성 구분에 효과적임을 보여주며, 성능이 디코더만의 결과가 아님을 뒷받침
✅ Data availability, Code availability
https://figshare.com/articles/dataset/TITANiAN_benchmark_result/26323582