💡 큰 주제: 발현 정제, Binding/Non-Binding
✅ 발현 정제란?
- 발현: 특정 단백질이 세포 내에서 얼마나 잘 생성되는가
- 정제: 복잡한 세포 내용물에서 원하는 단백질만 걸러내는 것
🔎 ProteinMPNN으로 설계한 de novo 단백질이 실제 세포에서 발현되고 정제되며,
실험적으로도 잘 접히고 항체와 결합한다면 성공
✅ 쉬운 설명
발현 + 정제는 ‘단백질 생산 공정’
1️⃣ 발현 (Expression) → 공장에서 단백질 "만들기"
내가 원하는 단백질 유전자를 세포 속에 넣음
세포는 이 유전자를 보고 단백질을 직접 생성함
쉽게 말해:
세포 = 공장, 단백질 = 제품
발현 -> “내가 설계한 단백질을 세포 공장에서 잘 만들 수 있는가?”
🖐 Q1. “서열을 아는데, 굳이 세포에 넣어야 해? 서열로 바로는 못 만들어?”
서열만으로는 실제 단백질을 만들 수 없음
서열은 ‘설계도’일 뿐이고, ‘단백질’은 물리적인 물질
세포에 서열 유전자를 넣어야
⟶ 리보솜이 해석해서 아미노산을 결합하고
⟶ 단백질을 물리적으로 합성해줌
🖐 Q2. “그럼 그 단백질 만들 세포는 어떻게 정해?”
목적 + 단백질 성질 + 실험 환경에 따라 적절한 host 세포를 선택
| Host 세포 | 특징 |
|---|---|
| E. coli | 값싸고 빠름, 단순 단백질, 실험용 초안 |
| Yeast | 진핵생물, 당화 일부 가능, 구조 단백질 |
| CHO (Chinese Hamster Ovary) | 인간 단백질 유사한 처리, 의약용 항체 |
| HEK293 | 사람 세포, 정확한 접힘, 고급 기능 단백질 |
2️⃣ 정제 (Purification) → 그 단백질만 깔끔하게 뽑아내기
세포 속에는 단백질뿐만 아니라 수천 가지 다른 물질도 같이 있음
그래서 내가 만든 특정 단백질만 선택적으로 뽑아내야 함
이를 위해 정제 기술 사용 (필터처럼 작동)
정제 -> “수많은 물질 중 내가 원하는 단백질만 깨끗하게 뽑을 수 있는가?”
✅ 실제 실험에서 발현 정제란?
ProteinMPNN으로 설계한 새로운 단백질(서열)을 실제로 써먹으려면:
그 단백질이 세포에서 만들어져야 함 (→ 발현)
다른 물질들과 섞이지 않고 깨끗하게 분리돼야 함 (→ 정제)
실험에서 접힘이 안정적이고 항체랑 잘 결합해야 함 (→ 성공)
🖐 Q1. “단백질을 뽑아내서 접힘이나 결합은 어디서 테스트해?”
정제한 단백질을 가지고 실험실에서 in vitro 분석
-
접힘 확인 (Folding, Stability 테스트)
SDS-PAGE + Western blot → 단백질 크기 및 유무 확인
Circular Dichroism (CD) → 이차 구조 확인
DSF (Differential Scanning Fluorimetry) → 열 안정성 -
결합 확인 (Binding Affinity, Specificity)
SPR (Surface Plasmon Resonance) → KD 측정
ITC (Isothermal Titration Calorimetry) → 결합 열량 측정
ELISA → 항원-항체 결합 여부
BLI (Biolayer Interferometry) → 항체 결합 정량 분석
🖐 Q2. “단백질 접힘(Folding)이라는 건 뭐야?”
단백질 접힘(Folding):
아미노산 서열이 정확한 3차원 구조로 접혀서 제 기능을 할 수 있는 형태가 되는 것을 의미
단백질은 구조(3D 구조)가 기능을 결정
-> 아미노산이 직선으로 줄줄이 있는 상태는 무의미, 접혀야 기능을 함
잘못 접히면 기능을 잃거나 독성이 생김 (예: 알츠하이머도 잘못 접힌 단백질이 원인)
✅ Binding / Non-Binding 이란?
두 생체분자(ex. 단백질-리간드, 단백질-펩타이드, 항체-항원 등) 가 결합(Binding)하는지 여부
-
1️⃣ protein:
아미노산이 연결된 고분자 생체분자
펩타이드보다 더 크고 복잡 -
2️⃣ peptide:
단백질보다 짧은 아미노산 사슬(50개 미만)
작은 단백질로 생각, 독립적인 생물학적 기능 가지기도 함 -
3️⃣ Ligand:
단백질에 결합하는 작은 분자
Receptor에 결합하여 기능 조절 -
4️⃣ Receptor:
Ligand를 인식하고 결합하는 단백질
세포 내부 반응을 유도 -
5️⃣ Antigen(항원):
면역 반응을 유발할 수 있는 분자(ex. 바이러스, 세균)
항체에 의해 인식됨 -
6️⃣ Antibody(항체):
항원을 인식하고 결합하는 Y자형 면역 단백질
항원을 중화하거나 면역 반응 유도 -
7️⃣ Binding:
두 분자가 특정 조건에서 물리적/화학적으로 상호작용하며 결합
이를 바탕으로 결합 예측 모델을 train 하거나, Docking 시뮬레이션 수행 -
8️⃣ Non-Binding:
두 분자가 결합하지 않음
ML에서 Negative Data로 중요하게 쓰임
💡 1. 포항공대와 협업
LLaMA 3.2를 finetuning(LoRA, QLoRa 통해)
✅ LLaMA 3.2 란?
Meta에서 개발한 오픈소스 LLM 시리즈
최신 버전은 LLaMA 3이며, 2024년 4월에 공개됨
성능은 GPT-3.5~4급이고, 공개된 파라미터는 8B, 70B 버전이 있음
🖐 Q1. finetuning 기법 중 LoRA는 왜 필요한가?
GPT나 LLaMA 같은 LLM은 수십억 개의 파라미터를 가짐
→ 전부를 fine tuning하면 메모리, 시간, 비용이 엄청나게 듬
→ 대부분의 NLP 작업에서는 모델 전체를 학습할 필요가 없음
🔎
기존 fine tuning의 경우 LLM을 업데이트하려면 각 개별 매개 변수의 업데이트 작업이 필요이는 수십억 개의 매개 변수를 fine tuning 하는 것을 의미 -> 방대한 컴퓨팅 시간과 비용이 소요
교사와 학급을 떠올려 보세요. 이 학급은 일 년 내내 수학을 배웠습니다.
시험 직전에 교사가 긴 나눗셈의 중요성을 강조합니다. 시험 도중에 많은 학생이 긴 나눗셈에 지나치게 몰두한 나머지 다른 문제에서 똑같이 중요한 수학 방정식을 잊어버렸습니다.
이는 기존 fine tuning에서 과적합으로 인해 LLM에 발생할 수 있는 결과입니다.
과적합: AI 모델이 일반적인 학습 데이터 외에 '노이즈', 즉 불필요한 데이터를 학습하게 되는 것을 의미
💡 LoRA는 AI모델의 일부만 변경
🔎
AI모델이 모든 것을 다시 학습하는 대신, 필요한 부분만 골라서 학습마치 학생이 이미 알고 있는 내용을 복습하지 않고, 새로운 내용만 공부하는 것과 같습니다.
그래서 작은 노력과 시간으로 훨씬 많은 것을 배울 수 있게 됩니다.
✅ LoRA(Low-Rank Adaptation)란?
LoRA는 기존 모델 파라미터를 freeze 하고,
일부 레이어에만 작은 저차원(=low-rank) 행렬을 학습시켜 fine tuning 하는 방법
기존 모델 W을 그대로 두고, 여기에 추가적인 변화만 압축형태로 학습함
A, B는 rank가 낮은 작은 행렬 (즉, 학습 파라미터 수가 매우 적음)
r은 low-rank (예: r=4, 8 등)
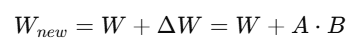
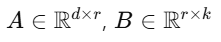
| 장점 |
|---|
| GPU 메모리 절약 (몇 GB 수준으로 fine-tuning 가능) |
| 빠른 학습 |
| 원래 모델 성능 보존 (base model은 그대로) |
🔎 기존 가중치 행렬
LoRA 저차원 행렬
전체 가중치 행렬에 적용
💡 결론
LoRA는 기존 weight W를 고정(freeze)하고,
새로운 저차원 행렬 ΔW=A*B만 학습함
하지만 최종 계산은 항상 W+ΔW로 이루어지기 때문에,
결과적으로 모델의 출력을 바꿀 수 있다
=> fine-tuning 효과가 난다.
전체 weight를 직접 수정하는 대신,
“덧붙이는 형태로 수정 방향을 제시하는 보조 weight”만 학습하는 것.
기존 weight W: 경험 많은 선생님
LoRA 행렬 A*B: 새로운 정보나 보정
결과적으로 출력은 바뀐다
→ 학습은 했고, 효과는 냈고, 비용은 적게 들었다
여기서 학습되는 건 AB뿐이지만, 모델 출력 전체는 바뀐다
그래서 이걸 통해 gradient가 흘러가고 loss가 줄어든다
🖐 어떻게 fine-tuning이 된다고 말할 수 있는가?
1️⃣ output이 바뀜: W+AB는 기존 W보다 다른 출력을 냄
2️⃣ gradient가 흐름: A, B는 trainable → 역전파 가능
3️⃣ 학습 대상은 작지만 강력함: 작은 보정만으로도 충분히 성능 향상 가능
4️⃣ 기존 knowledge는 유지됨: W는 pretraining 결과 그대로
🔎 예시
기존 모델은 잘 작동하지만 Edit Distance 10 같은 어려운 샘플은 오답을 냄
LoRA는 이런 샘플들에 대해 A*B를 학습
→ 기존 W의 결과를 살짝 밀어주듯 조정결과적으로 모델은 Edit 10 데이터에 대해서 더 나은 출력을 냄
→ 하지만 기존 knowledge는 유지됨
🖐 어디에 적용되는가?
LoRA는 주로 Transformer의 attention layer에만 적용
MultiHeadAttention:
- Q_proj (query)
- K_proj (key)
- V_proj (value)
- O_proj (output)
이 중 Q_proj와 V_proj에만 LoRA 적용 (논문에서 이 방식이 가장 성능이 좋았음)
🛑 주의:
1️⃣ 적용 대상:
기존 Transformer 모델의 Attention Layer 중 Query(Q), Value(V)
2️⃣ 문제: TensorFlow에는 peft 같은 LoRA 자동화 도구 존재 X
-> LoRA와 유사한 구조를 직접 구현해야함
기존 MultiHeadAttention Layer의 query, value projection에 대해서
W를 고정시키고, A * B 구조의 low-rank delta만 학습하는 구조를 추가로 만들어 MultiHeadAttention 출력에 더해주는 방식으로 구현
3️⃣ 그 후 문제:
기존 모델을 학습 시킬때, LoRA 관련 layer를 미리 만들어 LoRA는 turn off 한 상태로 학습 시킨게 아니라면,
모델이 학습된 후 그 후에 LoRA를 turn on 해서 학습시키는 것이 불가능
✅ QLoRA
🖐 Q1. 왜 QLoRA가 등장했는가?
LoRA도 메모리 절감이 되긴 하지만,
LLaMA-13B나 65B 모델은 여전히 너무 커서 학습 자체가 어려움.
→
그래서 QLoRA는 여기에 "양자화(quantization)"를 결합
QLoRA는 결국 기존의 LoRA에 새로운 quantization을 더한 형태
베이스 모델인 PLM의 가중치를 얼리고(frozen)
LoRA 어댑터의 가중치만 학습 가능하게(trainable)하는 것은 LoRA와 동일
frozen된 모델의 가중치가 '4비트로 양자화'되었다는 정도가 다른 점
장점:
- GPU 메모리 사용량 매우 적음 (24GB GPU로 65B 모델 fine-tuning도 가능)
- 성능이 Full Fine-tuning 에 근접 (실제 논문 기준으로 거의 손실 없음)
🖐 Q2. 모델을 양자화한다는 것이 뭔가요?
1️⃣ 양자화(Quantization)란?
실수(32bit float)로 표현된 값을 더 작은 정수(예: 4bit int)로 근사하여 표현하는 것
매우 복잡하고 정밀한 매개 변수(수많은 소수와 대량의 메모리)를 더 작고 더 간결한 매개 변수(더 적은 소수와 더 적은 메모리)로 압축하는 것을 의미
훨씬 적은 메모리로 행렬을 양자화할 수 있는 새로운 데이터 유형인 4비트 NF4(NormalFloat)가 사용됨
모델을 양자화하면 크기가 작기 때문에 fine tuning이 훨씬 더 수월
원래 모델 weight (float32):
𝑊= [0.234, −1.562, 3.94]양자화 후 weight (int4):
𝑊= [7, −12, 31]
이렇게 바꾸면, 메모리 사용량이 8배 줄어듦 (float32 → int4)
연산 속도도 빨라짐
2️⃣ QLoRA는 어떤 양자화를 쓰는가?
QLoRA는 4-bit 양자화 중에서도 특히 NF4 (NormalFloat4) 방식 사용
NF4 (NormalFloat4): float32 값을 4-bit로 근사하지만, 일반 정수보다 더 정밀하게 표현 (비선형 mapping 사용)
Double Quantization: 한 번 더 양자화해서 scaling factor를 압축 저장
Scale Factor: 정수 값(int4)만으로는 원래 weight를 정확히 표현할 수 없기 때문에,
이를 복원하기 위한 비율(스케일 값)을 함께 저장하는 값
🖐 Q3. Double Quantization 이 뭔가요?
Double Quantization은 양자화를 한 번 더 수행하는 기법
기존 모델의 매개변수

양자화된 모델의 매개변수

양자화할 때 매우 작은 일부 데이터는 압축 중에 손실될 위험 존재
분홍색 매개 변수 1개는 매개 변수에서 매우 작은 부분을 차지하여 압축된 버전으로 이어질 만큼 충분한 데이터를 대표하지 않기 때문에 누락됨
QLoRA에서는 모델 가중치를 양자화할 때 사용하는 양자화 상수(quantization constant)를 추가로 양자화하여 저장 공간을 더욱 줄임
이 방식은 양자화된 가중치뿐만 아니라, 가중치를 양자화할 때 생성되는 양자화 상수도 효율적으로 저장함으로써 모델 전체의 메모리 사용을 최소화
🔎 예시
원래 weight:
W = [0.0, 0.25, 0.5, 0.75, 1.0] → float32int4로는 정수만 표현되니까 이렇게 근사:
W_q = [0, 1, 2, 3, 4] → int4 (4-bit)그런데 우리가 04를 진짜 0.01.0로 해석하게 하려면?
→ 스케일링 계수 s 필요Dequantized value=s×W_q
여기서 s=0.25 이면:[0, 1, 2, 3, 4] × 0.25 = [0.0, 0.25, 0.5, 0.75, 1.0]
요소 의미 W_q 정수로 양자화된 weight (예: int4) scale 그 정수를 실제 값처럼 해석하기 위한 변환 비율 W_real ≈ scale × W_q 이게 최종적으로 계산에 쓰임
💡 즉,
“스케일 값이 너무 많아도 메모리를 차지하니까, 그 scale factor마저 한 번 더 양자화하는 것”
왜 필요하냐면?
원래 weight 10만 개 → int4로 양자화 OK
그런데 scale도 10만 개면 → float16 × 10만 개 => 메모리 부담
그래서 scale도 압축 → scale 벡터도 int8 or int4로 압축
float32 weights
↓ int4
W_q + scale (float16) ← 1차 양자화
↓ int8
quantized scale ← 2차 양자화 = Double Quantization
[ 원래 W (float32) ]
↓ 양자화 (NF4)
[ W_q (int4) + scale ]
↓ LoRA 추가
[ W_q * scale + A @ B ]
↓ forward 계산은 이걸로
💡 정리:
QLoRA는
1️⃣ 기존 가중치를 4bit로 양자화한다
2️⃣ 그걸 float처럼 쓰기 위해 scale factor를 만든다
3️⃣ scale이 너무 많으니 그것도 한 번 더 양자화한다 (Double Quantization)
🖐 Q4. 그 다음 LoRA를 붙일 땐
두 번 양자화된 값에 바로 LoRA를 붙이는가?
아니면 원래 값으로 복원(dequantize)한 후 LoRA를 붙이는가?
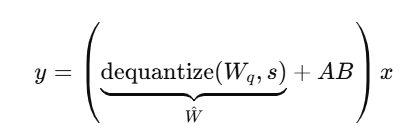
QLoRA는 양자화된 weight를 계산 시점에 "복원(dequantize)"해서,
그 복원된 값에 LoRA residual을 더하는 방식으로 작동
𝑊𝑞= 4-bit 양자화된 가중치
s = (양자화된) scale factor
^𝑊= 계산에 사용되는 복원된 float형 weight
A*B = 학습되는 저차원 residual
✅ 왜 “복원 후 더하기”가 맞는가?
LoRA는 기본적으로 float 연산 기반이기 때문
A*B는 저차원이긴 해도 여전히 float16 or float32 값
LoRA residual을 int4에 붙이면 precision mismatch로 의미 없음
따라서, 항상:
(1) 복원 → (2) LoRA 추가 → (3) 계산 의 순서로 작동
🛑 주의
문제:
QLoRA는 현재 HuggingFace + PyTorch 생태계에 맞춰 개발되어 있으며,
다음 라이브러리 기반으로 작동:
1️⃣ transformers (모델 정의 및 관리)
2️⃣ peft (LoRA/QLoRA 적용 및 관리)
3️⃣ bitsandbytes (4bit 양자화 및 optimizer 지원)
QLoRA는 보통 HuggingFace LLM 모델에 적용되므로, 커스텀 CNN/Transformer 구조에는 직접 적용은 어려움
HuggingFace에서 불러온 모델이 아니라 직접 만든 PyTorch 모델은
→ QLoRA의 양자화 적용은 현실적으로 어려움 (bitsandbytes는 HF 모델 전용 구조)
💡 그 후, ProteinMPNN 을 통해 자체 서열 생성하여, 새로운 단백질 생성
-> De novo 방식 + 발현까지 좋도록
✅ ProteinMPNN이란?
-
단백질 구조(3D)를 입력으로 하여 해당 구조에 맞는 아미노산 서열을 생성하는 딥러닝 모델
-
구조 기반 서열 설계 (structure-conditioned sequence design) 에 특화
-
Transformer와 GNN을 결합한 형태(MPNN, Message Passing Neural Network)의 모델로,
입력 단백질의 backbone 구조를 보고, 이를 안정적으로 구성할 수 있는 아미노산 서열을 예측
단백질 구조를 그래프, 노드는 아미노산 잔기로 봄
🔹 즉, 단백질 구조가 주어졌을 때, 그 구조를 잘 형성할 수 있는 서열을 역으로 생성해주는 모델
✅ ProteinMPNN의 기능들은 무엇이 있는가?
1️⃣ 구조 기반 서열 디자인
- 단백질 3D 구조를 입력으로 받아, 해당 구조를 형성할 수 있는 가능성 높은 서열을 생성
- 특히 backbone 정보만 있어도 작동하여 AlphaFold나 RosettaFold로 예측한 구조를 활용할 수 있음
2️⃣ de novo 단백질 디자인
- 전혀 존재하지 않던 새로운 단백질을 구조를 먼저 설계하고, 그 구조에 맞는 서열을 생성할 때 사용
3️⃣ Mutant 설계
- 단백질의 특정 위치에서 변이(mutation)를 유도
4️⃣ Site-specific redesign
- 단백질의 특정 위치에서 부분적인 서열만 고정하고 나머지를 설계
예) 항체 CDR 부위만 설계하고 framework는 고정
🖐 입력 구조는 어떻게 정하나?
1️⃣ AlphaFold/RoseTTAFold 등으로 예측한 구조
이미 서열이 있는 경우, 그걸 AlphaFold에 넣어서 구조를 예측
->
그 구조를 ProteinMPNN에 넣어서 서열을 다시 재설계
->
이렇게 하면 구조를 더 안정하게 형성할 최적의 서열을 만들 수 있음
예: 기존 단백질 서열이 잘 접히지 않거나 발현이 안 될 때 → 구조만 유지하면서 서열을 재설계
2️⃣ 구조를 직접 설계해서 넣는 경우 (도안처럼)
실험자나 연구자가 원하는 기능이나 모양을 가진 구조를 먼저 구상하거나 생성함
->
Rosetta, AlphaFold hallucination, trRosetta, AF2StructDiffusion 등의 생성모델들은 단백질 서열 없이도 3D 구조를 생성할 수 있음
즉, 뼈대만 “디자인”할 수 있다는 뜻
->
이때는 서열이 없음
→
이 구조(즉, backbone 좌표들)를 ProteinMPNN에 넣으면, 그 구조로 잘 접히는 아미노산 서열을 생성
🖐 ProteinMPNN 출력 서열은 어디에 쓰나?
그 서열이 실제로 접히는지 확인 → AlphaFold로 검증
만들어진 서열을 다시 AlphaFold에 넣고
→ 구조 예측 결과가 원래 도안한 구조와 비슷하면 접힐 가능성 높음
논문에 따르면 ProteinMPNN으로 재설계한 서열이 기존 네이티브 서열보다 AlphaFold로 예측 시 더 정확하고 안정적으로 예측됨
✅ ProteinMPNN 사용법 익히기
https://neurosnap.ai/service/ProteinMPNN -> ProteinMPNN 논문
https://github.com/dauparas/ProteinMPNN -> ProteinMPNN github
https://neurosnap.ai/service/ProteinMPNN -> ProteinMPNN 온라인 플랫폼(이걸로 사용법 감 잡기)
✅ De novo protein design란?
-
기존 자연 단백질 서열을 기반으로 하지 않고, 완전히 새롭게 단백질 구조 또는 서열을 설계하는 것
-
목표: 특정 기능(결합, 효소 활성 등)을 갖는 새로운 단백질을 만드는 것
💡 목표: 단백질 발현 향상("발현"이 목표-> 안정적인 단백질)
+ 항체 바인딩유지(affinity는 유지정도)
mouse 관련 단백질로 실험-> 추후 humanization이 목표
✅ 단백질 발현 향상이란?
세포 내에서 단백질이 더 많이, 안정적으로 생성되도록 서열이나 조건을 최적화하는 것
- 발현이 잘 되는 서열은 일반적으로 hydrophobicity, 안정성, folding kinetics 등이 좋음
- 발현량이 높고 misfolding이 적을수록 실험 성공률이 높아짐
- ProteinMPNN의 디자인을 통해 발현성이 떨어지는 서열을 발현이 잘 되도록 개선 가능
1️⃣ Hydrophobicity (소수성)
-
물을 싫어하는 성질
-
단백질에서의 의미: 아미노산 중에서 물을 싫어하는 소수성(amino acids like Leu, Val, Ile 등)이 많으면, 단백질 내부로 숨어버리려고 함
-
문제점: 지나치게 많은 소수성 잔기가 표면에 드러나 있으면 misfolding이나 aggregation(응집)을 유발하여 발현이 잘 안 될 수 있음
2️⃣ Misfolding
-
단백질이 원래 의도한 3차원 구조로 접히지 못하고, 잘못된 형태로 접히는 것
-
단백질은 아미노산 서열에 따라 정확한 구조로 접혀야 제 기능을 할 수 있음
-
하지만 구조가 잘못 접히면:
기능을 못 하거나
세포 내에서 분해되거나
오작동을 일으키는 단백질이 될 수 있음
ex) 알츠하이머병에서 β-아밀로이드 단백질이 잘못 접혀서 병을 일으킴
3️⃣ Aggregation (응집)
-
여러 단백질들이 엉겨붙어서 큰 덩어리(aggregate)를 형성하는 것
-
일반적으로 misfolding된 단백질들이 드러난 소수성 표면끼리 붙으면서 발생
-
Aggregation이 일어나면:
단백질이 비가용성(불용성)이 되어 실험에 사용할 수 없음
세포 독성 유발
단백질 발현량은 높아도 기능을 하는 단백질은 거의 없음
➡️ 그래서 발현 잘 되는 단백질은 소수성 아미노산이 내부에 잘 감춰져 있고, 표면에는 친수성아미노산이 배치된 경우가 다수
4️⃣ 안정성 (Stability)
-
단백질이 오래 유지되며 쉽게 변하지 않는 정도
-
열 안정성 (Thermal Stability)이나 화학적 안정성 등을 포함
-
구조적으로 안정한 단백질은 잘 접히고(잘 folding 되고), 변형이 적고, 긴 시간 동안 기능을 유지함
➡️ 단백질 발현 후 안정성이 낮으면 변성되거나 분해되어 원하는 실험을 하기 어려워짐
5️⃣ Folding kinetics (접힘 속도)
-
단백질이 자연적으로 제대로 접히는 속도
-
단백질은 1차 서열(아미노산 배열)이 주어지면 스스로 3차원 구조로 접히는데, 이 접힘이 느리거나 잘못되면 발현 문제 발생
-
특히 고속 발현 시스템(E. coli 등)에서 빠르게 접히는 서열이 발현 성공률이 높음
-
folding이 빠르고 효율적인 단백질은 세포 내에서 체내 품질관리 시스템(Ubiquitin 등)에 의해 제거되지 않고 기능할 수 있음
6️⃣ 고속 발현 시스템 (High-expression system) 이란?
-
단백질을 빠르고 많이 생산할 수 있도록 설계된 생명체 또는 세포 시스템
-
대표적인 예:
E. coli (대장균)
Yeast (효모)
CHO cell (중국 햄스터 난소 세포)
HEK293 (인간 신장 세포)
-> 이 중에서 E. coli는 가장 많이 사용되는 대표적인 고속 발현 시스템 -
왜 E. coli를 고속 발현 시스템으로 쓸까?
| 장점 | 설명 |
|---|---|
| 빠른 성장 | 20~30분에 1번씩 증식 → 하루 만에 많은 양 가능 |
| 저렴한 배양 | 영양분, 장비가 저렴함 |
| 유전 조작 쉬움 | 플라스미드 삽입 등 유전자 조작 간편 |
| 단백질 발현량 높음 | 특정 단백질을 하루 만에 대량 생산 가능 |
| 단점 | 설명 |
|---|---|
| 복잡한 단백질 접힘 불가 | E. coli는 진핵생물이 아니라 정교한 folding, glycosylation 등 처리 못 함 |
| misfolding / aggregation 많음 | 소수성 단백질이나 큰 단백질은 잘못 접히거나 inclusion body로 뭉침 |
- 그래서 발현 성공을 높이려면?
ProteinMPNN 같은 도구를 통해 잘 접히는 서열로 설계하고,
aggregation을 줄이는 방향으로 설계하면
E. coli 같은 고속 시스템에서도 안정적인 발현이 가능
7️⃣ 체내 품질관리 시스템이란?
- 세포는 생존을 위해 오작동하거나 잘못 접힌 단백질들을 감지해서 파괴하는 시스템을 갖추고 있음, 이를 판단하고 제거하는 시스템
✅ affinity란?
항체나 리간드가 표적 단백질에 얼마나 강하게 결합하는지를 나타내는 지표
-
Kd (dissociation constant)로 측정되며, 값이 낮을수록 강한 결합(음수값이 좋은 값)
-
바인딩 능력을 유지한 채로 다른 부분을 최적화하는 것이 중요
-
ProteinMPNN으로 서열을 바꿀 때, 결합 부위는 고정하고 나머지만 설계하여 affinity 유지 가능
✅ mouse 관련 단백질로 먼저 실험하는 이유는?
-
모델 동물 중 가장 많이 사용됨
→ 유전정보, 생리학적 특성 등이 잘 알려져 있음 -
실험 환경이 잘 구축됨
→ 동물실험 및 항체 생성 시스템 등 -
사람 항체 개발 전 테스트용
→ 항체 효능이나 안정성 등을 먼저 확인 가능
✅ Humanization 이란?
비인간 항체 또는 단백질의 일부를 사람의 서열로 바꾸는 과정
-
면역원성을 낮추고 인체에 투여 가능한 치료제로 만들기 위한 과정
-
보통 CDR Region은 유지하고 framework만 human 서열로 변경
-
humanization 이후에도 기능과 구조 안정성 유지가 매우 중요 → 이를 위해 구조 기반 모델(예: AlphaFold + ProteinMPNN 등)이 도움됨
💡 affinity는 AlphaFold, Schrödinger 사용하여 예측중
AlphaFold, Schrödinger: 단백질 구조 및 상호작용 예측에서 중요한 도구
✅ AlphaFold 1이란?
Google DeepMind가 개발한 단백질 구조 예측 모델
💡 AlphaFold 1이 MSA, DNN, ResNet을 활용하는 이유
1️⃣ AlphaFold 1은 단백질의 유사한 서열들을 찾아내어 정렬한 다중 서열 정렬 (Multiple Sequence Alignment, MSA)을 학습 데이터로 구성
2️⃣ MSA 데이터는 서열 내 각 아미노산 간의 공진화(co-evolution)에 대한 정보를 포함
-> 진화론적 관점에서도 단백질 구조 형성에 대한 힌트를 제공
🖐 단백질 구조 예측에서의 co-evolution
단백질 서열 상의 아미노산 위치들이 서로 진화적으로 연관된다는 것
AlphaFold 같은 단백질 구조 예측 모델에서, 공진화 정보를 이용해 서로 가까운 3차 구조 상의 아미노산 쌍을 추론할 수 있음
단백질이 진화하면서 어떤 아미노산 위치 A가 변할 때, 그 위치와 상호작용하는 다른 위치 B도 함께 변하는 경향 존재
🔎 ex) A와 B가 공진화하는 경우 → 실제 3차 구조에서도 가까운 거리일 확률이 높음 → 접촉 예측에 활용
3️⃣ AlphaFold 1은 ResNet의 구조를 차용한 DNN(Deep Neural Network)으로 구성된 딥러닝 모델
ResNet을 구성하는 CNN 은 이미지나 MSA 데이터와 같은 2차원 데이터의 공간 정보를 효과적으로 학습
앞서 언급한 것처럼 MSA 데이터는 주어진 단백질 서열 내 아미노산 간의 진화론적인 상관관계를 2차원 공간에 함축한 형태
따라서 CNN 기반의 신경망은 알파폴드 1에 적합한 구조
💡 AlphaFold 1이 MSA 데이터를 활용 하는 과정
1️⃣ AlphaFold 1은 DNN을 통해 MSA 데이터로부터 각 아미노산의 중심이 되는 탄소 간의 거리와 뒤틀림각의 분포를 학습
2️⃣ 만약 3차원 공간에서 각 아미노산 간의 거리와 뒤틀림각의 분포를 알 수 있다면, 이를 기반으로 단백질의 구조의 안정성을 평가하는 통계적 퍼텐셜 (statistical potential) 함수를 구성할 수 있음
3️⃣ AlphaFold 1은 여기에 Rosetta 프로그램을 통해 단백질 구조의 유효성을 평가하는 또 다른 퍼텐셜 함수를 더하여 최종 퍼텐셜 함수를 구성
4️⃣ AlphaFold 1은 예측한 뒤틀림각 분포로부터 gradient descent을 이용하여 가장 안정한 형태를 이루는 아미노산들의 뒤틀림각 조합을 찾음
이러한 과정을 거쳐 AlphaFold 1은 최종 단백질 구조를 예측
💡 쉽게 보는 AlphaFold 1의 단백질 구조 예측 과정
1️⃣ MSA 데이터를 활용한 학습 데이터 구성
2️⃣ CNN 네트워크 구조를 차용한 DNN 구축
아미노산들 간의 거리 및 뒤틀림각 분포를 학습
3️⃣ 네트워크로부터 예측된 아미노산 간의 거리 및 뒤틀림각 분포를 이용한 퍼텐셜 함수 구성
4️⃣ 3차원 구조를 아미노산들의 뒤틀림각으로 수치화 한 뒤 경사 하강법을 통해 퍼텐셜이 최소가 되도록 뒤틀림각 조합 최적화
✅ AlphaFold 2 란?
AlphaFold 2는 AlphaFold 1과 비교해 어떤 점이 달라졌고, 얼마나 성능 향상이 되었는가?
💡 MSA 기반 학습 데이터의 확장
AlphaFold 2는 AlphaFold 1에서 쓴 MSA 데이터를 더 확장하여 총 2가지의 학습 데이터를 구성
1️⃣ 첫 번째는 AlphaFold 1처럼 단백질 서열 데이터베이스를 탐색하여 구성한 MSA representation 데이터
이 MSA 데이터를 활용하여 단백질 구조 데이터베이스를 추가로 탐색
2️⃣ 두 번째는 단백질의 서열과 구조 간의 상관관계에 대한 정보를 제공하는 template을 기반으로 pair-representation 데이터를 구성
💡 CNN 기반 ResNet -> attention 기반 Evoformer 로 모델 구조의 변화
AlphaFold 2의 네트워크 구조: MSA를 더 효과적으로 사용하기 위한 Evoformer
1️⃣ Evoformer의 가장 중요한 핵심은 데이터 내의 구성 요소들 간의 상관관계를 고려하는 ‘attention’이라는 개념을 이용하는 것
2️⃣ Attention을 통해 MSA representation data 내에서는 서로 다른 단백질 서열들 간의 상관 관계를 추출 +
pair representation 내에서는 입력 단백질 내의 아미노산들 간의 상관 관계를 학습하여 효과적으로 함축된 또 다른 정보를 추출
3️⃣ 또한 Evoformer는 MSA & pair representation 사이에 정보를 교환하는 연산을 추가하여 두 입력 데이터가 서로를 반영하면서 업데이트 될 수 있도록 도움
💡 End-to-end 형태로 단백질 구조 예측 방식의 변화
1️⃣ AlphaFold 1 은
1. 네트워크를 통한 거리 및 뒤틀림각 분포 예측 →
2. 예측한 분포를 통해 구성한 퍼텐셜 함수의 최적화
라는 2개의 별도의 과정을 통해 단백질의 구조를 예측
2️⃣ AlphaFold 2는 입력 데이터로부터 최종 예측까지 하나의 네트워크로 이어지는 end-to-end 형태로 바로 단백질 구조를 예측
3️⃣ 이를 가능하게 해주는 것이 Evoformer 뒷단에 추가된 Structure module 네트워크
4️⃣ Structure module은 Evoformer를 통해 업데이트된 MSA & pair representation을 입력으로 받음
그리고 attention이 접목된 네트워크 구조인 Invariant Point Attention module (IPA module)을 통해 추가로 업데이트를 진행
이렇게 최종적으로 업데이트된 정보를 바탕으로 각 아미노산마다 유클리디언 변환 (Euclidean Transformation) 행렬을 예측
5️⃣ AlphaFold 2는 이러한 학습 과정에서 예측 데이터를 누적
6️⃣ 처음에는 모두 원점에서 시작됐던 단백질 내 아미노산의 좌표가 실제 단백질 구조의 위치로 이동
아미노산들의 좌표 정보 (backbone)가 업데이트되면, 이를 바탕으로 아미노산마다 원자들의 뒤틀림각을 예측하여 원자들의 좌표를 예측
7️⃣ 추가로 AlphaFold 2는 업데이트한 MSA & pair representation과 예측한 구조 정보를 다시 Evoformer와 Structure module의 입력 데이터로 넣어주는 recycling 과정을 가짐
이를 통해 전체 네트워크를 반복적으로 학습시켜 최종적으로 알파폴드 1보다 더 정확한 단백질 구조 예측 가능
✅ AlphaFold 3란?
-
기존 AlphaFold2보다 훨씬 더 다양한 분자 복합체 예측이 가능
-
단일 단백질뿐만 아니라, DNA, RNA, 리간드(소분자), 이온, 물 등을 포함한 복합체의 구조 예측 가능
-
2024년에 발표된 최신 버전으로, 기존의 구조 예측에서 복합체 상호작용 예측으로 확장
🖐 AlphaFold3 변화점
💡 1. 입력 데이터 구성 및 업데이트
1️⃣ AlphaFold 2는
구조를 예측하고자 하는 단백질 서열에 대한 진화론적 힌트를 주는 MSA 데이터 +
구조적인 힌트를 제공하는 template 데이터를 기반으로 형성한
pair representation 데이터
이 두 가지 데이터를 입력으로 받음
이때 AlphaFold2는 오직 단일 단백질에 대한 구조를 예측하는 모델이기 때문에
이 두 데이터의 기본 단위는 단백질 서열의 구성 요소인 아미노산
2️⃣ AlphaFold 3는
예측하고자 하는 대상이 단백질뿐만 아니라 핵산과 리간드도 포함
입력 서열을 각 구성 성분의 유형에 따라 서로 다른 입력 단위로 표시
즉, 입력 서열 내에 단백질은 아미노산, 핵산은 뉴클레오타이드, 그리고 리간드는 원자 단위로 표시하여 초기 입력 데이터 구성
이렇게 구성된 입력 데이터는 Input Embedder에서 각 요소가 합쳐진 복합체 단위로 업데이트를 진행
3️⃣ 서로 다른 단위를 가지는 입력 데이터를 어떻게 하나의 복합체로 통합한 뒤 업데이트할 수 있는가?
->
아미노산이나 뉴클레오타이드 단위들을 최소 입력 단위인 원자 단위로 쪼갠 뒤 연산을 수행
원자 단위로 입력 단위를 세분화하면 임의의 생체 분자 복합체가 입력으로 들어오더라도 원자라는 공통된 입력 단위를 토대로 전체 복합체를 표시할 수 있고 추후 하나의 일관된 연산을 적용 가능
이때 reference conformer라는 추가 데이터가 사용되는데,
이것은 아미노산이나 뉴클레오타이드와 같은 분자 단위의 입력을 원자 단위로 쪼갤 때 필요한 각 분자의 원자 구성 및 구조 정보를 제공
이렇게 재구성된 입력 데이터는 Input Embedder 네트워크 내에서 원자 단위의 연산을 통해 업데이트
이때 각 원자마다 상관관계(attention)를 고려하는 연산을 수행하기 때문에 이 연산을 AtomAttention이라고 부름
4️⃣ AtomAttention 연산 결과값은 다시 원래 입력 단위로 재구성되어 업데이트 된 입력 서열을 형성
이는 AlphaFold3가 AlphaFold2와 마찬가지로 MSA 및 template 데이터 사용하여 추가적인 업데이트를 진행하기 때문
MSA 와 template 데이터는 아미노산 혹은 뉴클레오타이드 단위로 표현되기 때문에 AtomAttention 연산 결과값 내에 원자 단위로 쪼개진 아미노산/뉴클레오타이드 표현을 다시 분자 단위 표현으로 합쳐주는 것
그리고 업데이트된 입력 서열에 외적 연산을 적용하여 pair representation을 형성한 뒤,
이를 순차적으로 Template module 과 MSA module을 통해 각각 template, MSA 데이터의 정보와 결합하여 업데이트
💡 AlphaFold3의 입력 데이터 구성과 업데이트 과정 요약
1️⃣ 입력 : 입력 서열
단백질은 아미노산, 핵산은 뉴클레오타이드, 그리고 리간드는 원자 단위로 입력 단위를 구성하여 초기 입력 데이터를 구성
2️⃣ 입력 데이터를 reference conformer 데이터를 활용하여 Input Embedder 내에서 원자 단위로 쪼갠 뒤 원자 단위로 상관관계를 고려하며 업데이트하는 AtomAttention 연산을 수행
3️⃣ AtomAttention 결과 업데이트된 single representation을 기반으로 pair를 형성한 뒤, Template & MSA module 내에서 template & MSA 데이터의 정보를 결합하여 업데이트
4️⃣ 출력 : 업데이트 된 single 및 pair representation
💡 2. 내부 모듈의 변화 : Evoformer에서 Pairformer로
1️⃣ AlphaFold2에서는 MSA 데이터와 pair representation 데이터를 Evoformer라는 네트워크를 통해 업데이트
Evoformer는 attention을 사용하여 MSA 내에서는 서로 다른 단백질 서열 간의 진화론적인 상관관계를 고려하고,
pair representation 내에서는 입력 단백질 서열 내의 아미노산 간의 상관관계를 고려하며 각 데이터를 업데이트
추가로 Evoformer는 연산 도중 MSA 와 pair representation 사이에 서로 정보를 한번씩 교환하는 연산이 존재하여
두 입력 데이터가 서로의 정보를 반영하면서 업데이트되도록 유도
2️⃣ AlphaFold3에서는 Evoformer와 유사한 Pairformer Network를 사용
Pairformer와 Evoformer간의 가장 큰 차이점: 입력으로 받는 데이터의 종류와 내부 연산
Evoformer가 입력 서열과 진화론적으로 유사한 다른 단백질 서열까지 포함된 MSA 데이터를 입력으로 받는 대신에,
Pairformer는 Input Embedder에서 업데이트된 입력 서열(single representation)을 받음
따라서 Evoformer와 달리
MSA 내부에서 서열 간 attention 연산과
MSA 와 pair representation 간의 정보 교환 연산이
각각 업데이트된 서열(single representation) 내의 요소들끼리의 attention 연산과
pair에서 single representation으로의 하나의 정보 교환 연산으로 간소화됨
이때 Pairformer에서 빠진 MSA와 pair representation간의 정보 교환 연산은 MSA module로 대체
💡 Pairformer 내의 데이터 흐름과 연산 요약
1️⃣ 입력 : Input Embedder에서 업데이트한 single & pair representation
2️⃣ Pair representation 내에 요소들끼리의 attention을 통한 연산을 통해 업데이트
3️⃣ 업데이트된 pair representation의 정보를 single로 전달
4️⃣ pair의 정보를 받은 single representation을 업데이트
5️⃣ 출력 : 업데이트된 single & pair representation
Pairformer는 Evoformer를 토대로 입력으로 받는 데이터의 크기
(여러 서열로 이루어진 MSA 데이터에서 입력 서열로만 이루어진 single representation으로) 와
내부 연산 (MSA ↔ pair 정보 교환이 pair → single로)이 간소화 된 네트워크라고 할 수 있음
💡 3. 새로운 구조 예측 네트워크, Diffusion
1️⃣ AlphaFold2는 Evoformer에서 업데이트된 MSA & pair representation을 기반으로
Structure module 을 통해 최종적으로 단백질의 구조를 예측
2️⃣ Structure module은 먼저 입력으로 받은 MSA & pair representation으로부터 ->
아미노산들의 유클리디언 변환 (Euclidean Transformation) 행렬을 예측하여
backbone의 위치를 구한 뒤 ->
각 아미노산 마다 뒤틀림각을 예측하여 ->
아미노산 내의 개별 원자들의 좌표를 예측해 내는 식으로 단백질 내 전체 원자들의 3차원 좌표를 예측
그리고 현재까지 예측한 단백질 구조 정보를 다시 Structure module의 입력으로 넣어주는 과정을 반복하여
단백질 내 원자들이 원점에서 점진적으로 실제 좌표값으로 이동하게 만듦
3️⃣ AlphaFold3는 생체 분자의 구조를 예측하기 위해 이미지 생성에 널리 쓰이는 AI 모델 중 하나인 Diffusion을 사용
4️⃣ Diffusion이란
원본 데이터(이미지)에 점진적으로 노이즈를 준 뒤,
그것을 제거하는 과정을 네트워크를 통해 학습시켜
최종적으로 완전한 노이즈에서 학습한 데이터와 비슷한
새로운 데이터를 생성할 수 있는 생성형 AI 모델
5️⃣ AlphaFold3에서는 이 Diffusion을 기반으로
노이즈에서 생체 분자 내의 원자들의 3차원 좌표를 생성하는
Diffusion module로 Structure module을 대체
6️⃣ 먼저 Pairformer의 출력인 업데이트 된 single & pair representation을 입력으로 받아서
Diffusion Conditioning 연산을 수행하여 각 원자의 3차원 내 공간적 조건을 계산
이때 single & pair representation 내에서 단백질/핵산의 단위는 아미노산/뉴클레오타이드이기 때문에 각 원자마다 조건을 할당하기 위해서
Input Embedder처럼 데이터 내 입력 단위를 모두 원자 단위로 쪼개주는 과정이 포함됨
7️⃣ 이렇게 계산된 공간적 조건들은 노이즈가 추가된 원자들의 3차원 좌표 정보와 결합하여 노이즈가 추가되기 전 올바른 3차원 좌표를 예측
8️⃣ AlphaFold3는 학습 시 생체 분자의 실체 3차원 구조 (Ground truth) 하나당 여러 개의 노이즈가 추가된 샘플을 만들어 내고
각 샘플을 Diffusion module의 입력으로 넣어줘서
Ground truth 내의 3차원 좌표들을 예측하도록 함
즉, Diffusion module은 학습 과정에서는 노이즈를 한 번만 제거하는 single step of the diffusion을 학습하는 것
9️⃣ 그러나 추론 과정에서는 single step of the diffusion을 열거하여
각 step마다 나온 노이즈가 제거된 좌푯값을 다시 Diffusion module의 입력으로 넣어 주는 과정을 반복
이것은 AlphaFold2에서 Structure module이 이전 구조 예측값을 입력으로 받으며 점진적으로 개선하는 과정과 비슷하며
이 과정을 논문에서는 mini-rollout이라고 언급
🔟 이러한 mini-rollout 과정을 통해 Diffusion module은 최종적으로 완전한 노이즈를 점진적으로 원자들의 3차원 좌표들로 변환할 수 있게 됨
💡 Diffusion module이 최종적으로 원자들의 3차원 좌표를 예측하는 과정 정리
1️⃣ 입력 : Pairformer에서 업데이트한 single & pair representation +
노이즈가 추가된 원자들의 3차원 좌표
2️⃣ 업데이트된 single & pair representation을 원자 단위로 세분화한 뒤
각 원자마다 3차원 공간적 조건을 담도록 업데이트
3️⃣ 업데이트 각 원자별 공간적 조건과 노이즈가 추가된 원자들의 3차원 좌표를 결합하여 노이즈가 제거된 원본 좌표값을 예측
4️⃣ 출력 : 노이즈가 제거된 원자들의 3차원 좌표
💡 4. AlphaFold3 정리
AlphaFold3는 AlphaFold2와 비교했을 때 크게 아래 3가지 요소들에서 특징적
1️⃣ 임의의 생체 분자 복합체를 입력으로 받을 수 있게 입력 서열의 구성과 업데이트 방식 변화
2️⃣ Evoformer보다 간소화된 Pairformer를 통해 singe & pair representation을 업데이트
3️⃣ 원자들의 3차원 좌표를 예측하기 위해서 생성형 AI 기술인 Diffusion을 접목
AlphaFold 참조 링크
https://hyperlab.hits.ai/blog/google-deepmind-alpha-fold-1-and-2
https://hyperlab.hits.ai/blog/AlphaFold3-Review
✅ 알파폴드 3의 기능에는 무엇이 있는가?
AlphaFold 2: 단일 단백질의 3차원 구조 예측 (PDB 수준 정확도)
AlphaFold-Multimer: 단백질-단백질 복합체 구조 예측 (복수 서열 입력)
1️⃣ 복합체 예측:
단백질–단백질, 단백질–DNA, 단백질–RNA, 단백질–리간드 등
거의 모든 유형의 단백질 기반 생체 분자 복합체의 3차원 구조를 정확히 예측
2️⃣ 결합 친화도 추정 (간접적으로):
정확한 binding affinity 수치 예측은 직접 하지는 않지만,
결합 인터페이스를 예측함으로써 구조 기반 에너지 분석 가능
-
Interface 분석
AlphaFold가 예측한 복합체 구조에서 단백질–단백질 접촉면의 크기나 결합된 형태의 안정성을 분석함 -
PAE Matrix 해석
결합 영역 사이의 PAE 값이 낮을수록 → 구조 예측 신뢰도 높음 → 결합이 안정적일 가능성 ↑
🖐 PAE (Predicted Aligned Error)
예측된 정렬 오차
→ AlphaFold가 두 아미노산 잔기 사이의 상대적 위치를 얼마나 정확히 예측했는지를 수치로 나타낸 것
-
구조 기반 도킹 scoring
AlphaFold 구조를 기반으로 Schrödinger의 Glide / MM-GBSA / FEP+ 등의 소프트웨어에 넣어 정확한 binding affinity 계산 -
AlphaFold 구조 + 에너지 계산 툴
AlphaFold → 구조 예측 → GROMACS, Rosetta, FoldX 등을 통해 결합 에너지 계산 가능
✅ 알파폴드 사용법 익히기
https://github.com/google-deepmind/alphafold3?tab=readme-ov-file
✅ 슈뢰딩거란?
- 미국의 제약소프트웨어 회사로,
정밀한 분자동역학(MD), 리간드 도킹, 결합 친화도 예측, 약물 물성 분석, 구조 기반 약물 설계(SBDD) 등의
컴퓨터 기반 약물 개발(CADD) 소프트웨어 플랫폼을 제공
✅ 슈뢰딩거의 기능에는 무엇이 있는가?
-
Glide: 고속 리간드 도킹 프로그램
단백질 표면에 리간드를 dock하고, binding affinity를 추정 -
Prime: 단백질 구조 모델링
Loop modeling, missing side chain 복원, point mutation 구조 생성
Ab initio 방식으로 변형된 단백질 구조 생성 가능
🖐 Loop Modeling
단백질에서 loop 영역은 α-helix나 β-sheet 같은 규칙적인 구조가 아닌, 불규칙하고 유연한 부분
실험 구조(PDB)에서 loop 영역이, 해당 PDB 구조에 안 잡히거나 결실(missing residues)이 생길 수 있음
Loop Modeling이 필요한 이유:
리간드나 파트너 단백질과의 결합에 직접 관여하는 결합 부위가 loop에 위치하는 경우가 많기 때문에, 정확한 구조 모델링이 중요
특히 항체에서는 CDR 루프가 항원을 인식하는 핵심 부위
그러나 루프는 유연해서 실험(PDB 구조)에서는 결정되지 않거나 누락될 수 있음
ex)
PDB 구조에선 110~120번 루프가 아예 없음 →
그런데 이 루프가 biding pocket을 형성함 →
Docking을 해도 결합 부위가 제대로 없거나 찌그러져 있음
특히 docking 시뮬레이션 시 loop 없으면 구조가 깨짐
현상 설명 루프 누락 단백질의 표면에 구멍이나 비정상적인 공간이 생김 도킹 오류 발생 리간드가 잘못된 위치에 결합하거나, 비정상적인 자세로 박힘 에너지 계산 오류 실제 결합보다 더 안정해 보이거나, 전혀 결합하지 않는 것처럼 나옴 구조 왜곡 루프가 없어서 단백질이 실제보다 덜 압축돼 보이거나 공간적 방해(Steric hindrance)가 생기지 않음
루프가 없으면 docking이 잘못됨 →
결과 해석도 잘못됨 →
binding affinity 예측 정확도 하락
ex)
원본 PDB: residue 120~130 loop가 결실됨
→ Prime 또는 MODELLER 등으로 해당 loop 부분을 자동 예측 및 생성
Loop Modeling 이 필요한 경우
| 상황 | 이유 |
|---|---|
| 결합 부위에 loop가 포함된 경우 | 정확한 포켓 형성 필요 |
| 항체–항원 결합 구조 | CDR 루프는 대부분 유연하고 PDB에 누락돼 있음 |
| AlphaFold 구조 사용 시 | 일부 루프의 정확도가 낮음 → 보정 필요 |
| 변이 실험 전 | 루프를 다시 모델링해서 안정성 및 binding 영향 분석 가능 |
🖐 Missing Side Chain
PDB 실험 구조에서는 종종 side chain이 전자 밀도 부족으로 인해 빠져 있을 수 있음
(특히 Lys, Arg, Glu 같은 긴 side chain)
왜 중요한가?
Hydrogen bonding, salt bridge, van der Waals contact 등은 side chain에서 주로 발생함
도킹 정확도 및 MD 에너지 계산에 큰 영향을 줌
🖐 Point Mutation 구조 생성 (점 돌연변이)
단백질 서열에서 특정 아미노산 하나를 다른 아미노산으로 바꾸는 것
사용 목적
돌연변이가 구조/기능에 미치는 영향 분석
예측된 돌연변이 구조를 기반으로 결합력 변화, 안정성 변화 등을 시뮬레이션
-
Desmond: 분자동역학(MD) 시뮬레이션
-
MM-GBSA: 결합 에너지 계산
Binding affinity 비교에 자주 사용 -
FEP+: 결합 친화도 예측
두 리간드 간 상대적 결합 친화도를 열역학적으로 정확하게 계산 -
QikProp: ADMET 물성 예측
리간드의 흡수(Absorption), 분포(Distribution), 대사(Metabolism), 배설(Excretion), 독성(Toxicity) 예측
✅ 슈뢰딩거 사용법 익히기
기업/학교 라이선스 보유 시 설치 가능
💡 2. 생명 팀원들 데이터 그래프 그려주기
-> 논문 데이터 분석 시에 사용 위해
-> R 미리 보고 오기
✅ R 이란?
통계 분석과 데이터 시각화에 특화된 프로그래밍 언어
생명과학, 바이오, 의학 분야에서 논문용 그래프 제작이나 통계적 유의성 검증 등에 많이 사용
✅ R의 기능에는 무엇이 있는가?
1️⃣ 통계 분석
2️⃣ 데이터 처리 및 정제
3️⃣ 데이터 시각화
✅ R 사용법 익히기
생명 팀원들 데이터 분석 시 적용 예시
팀원들 실험 결과를 CSV로 정리 → R로 불러오기
통계 검정 → 유의성 확인
ggplot2로 예쁜 그래프 제작 (논문 제출용 품질)
실제 사용은 GPT 로 돌려도 충분히 가능할 듯
💡 3. Randomforest / SVM 이용하여
환자, 비환자 Binding 정보 분석
U-MAP을 통해 Singel Cell 분석
✅ Randomforest 란?
여러 개의 Decision Tree(결정 트리)를 만들어서,
그 결과를 투표 혹은 평균 하여 최종 예측을 내리는 앙상블 학습(Ensemble Learning) 알고리즘
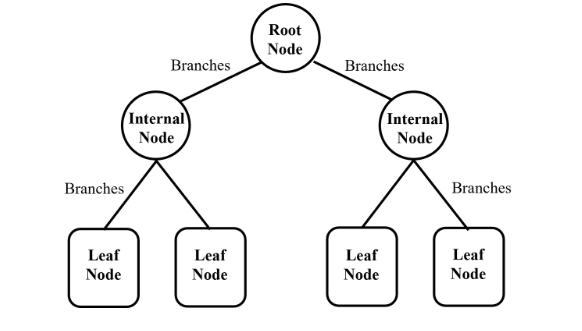
- 분류(Classification) → 다수결 투표
- 회귀(Regression) → 평균값 예측
🖐 앙상블 학습(Ensemble Learning)
여러 개의 모델을 결합해서 하나의 더 강력한 모델을 만드는 방법
1️⃣ 여러 트리는 어떻게 만들어지는가?
Bootstrap Sampling: 각 트리를 만들 때 데이터를 중복 허용하며 샘플링
Random Feature Selection: 각 노드를 나눌 때 모든 feature 중 일부만 랜덤하게 선택
->
이러한 랜덤성과 다수의 트리가 결합되어 과적합을 방지하고, 예측 성능이 향상
2️⃣ 하나의 트리는 어떻게 구성되는가?
ex)
100개의 샘플, 20개의 feature가 있는 데이터1️⃣ Bootstrap 샘플링:
트리 A: 100개 데이터 중, 중복 허용하여 100개 선택 → A만의 훈련 데이터
트리 B: 또 다른 무작위 샘플링 → B만의 훈련 데이터
->
각 트리는 서로 다른 데이터를 학습함
2️⃣ 노드 분할 시 Feature Subset 사용:전체 feature가 20개라면 각 노드에서 무작위로 5개만 뽑고, 그 중 최적 분할 찾음
->
이렇게 하면 트리마다 다른 기준으로 분기함 → 결과적으로 다양한 구조 생성됨
🔎 쉬운 예시
RandomForest = 여러 사람에게 의견을 물어보는 과정
같은 질문(예: 이 사람 환자일까?)을 여러 사람(트리)에게 물어보는데,
사람마다 다른 경험(훈련 데이터)과 다른 기준(랜덤 feature)으로 판단함
그리고 마지막에 투표해서 다수가 말하는 쪽을 선택함→ 여러 관점을 종합하기 때문에 과적합을 줄이고, 일반화 성능이 향상됨
🔎 활용 예시 (의료 데이터 기준)
입력 feature:
환자 데이터 (예: 유전자 발현값, 항체 바인딩 세기, 혈액 내 단백질 수치 등)타겟 label:
환자(1) vs 비환자(0)활용:
바인딩 특성을 기반으로 환자군을 예측 → 조기 진단 모델, 바이오마커 탐색 등에 활용 가능
✅ SVM(Support Vector Machine) 란?
데이터를 분류하는데 있어 두 클래스 간의
최대 margin(여백)을 확보하는 선형 또는 비선형 분리 경계를 학습하는 알고리즘
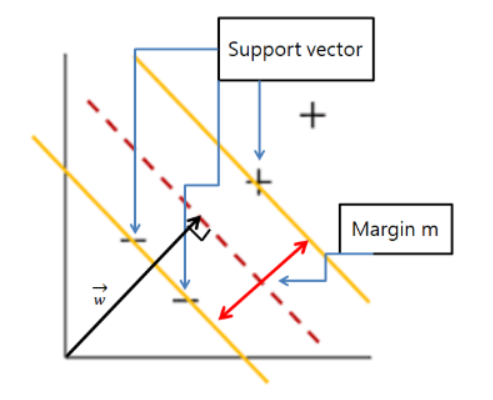
🔎 환자 분류 활용 예시
단백질 바인딩 정보가 feature일 때,
SVM은 이를 기반으로 결정 경계를 학습해 환자군과 비환자군을 구분할 수 있음
✅ 환자, 비환자 Binding 정보 분석이란?
환자와 비환자의 단백질-리간드 또는 항체-항원 Binding 정보를 기반으로,
면역 반응이나 질병 관련 특성 차이를 분석하는 것
🔎 예시
환자군에서 특정 epitope에 대한 항체 결합이 더 강함
비환자군에선 특정 TCR이나 BCR 클론이 결합하지 않음
->
이런 차이를 통해 바이오마커나 진단 기준을 설정 가능
🖐 Machine Learning 연계
위 정보를 feature로 사용해 Random Forest, SVM 등으로 질병 예측 모델을 만들 수 있음
🖐 Epitope (에피토프, 항원결정기)
항원(Ag)의 표면에서 항체 또는 T세포 수용체(TCR)가 인식하고 결합하는 특정 부위
-> 면역 시스템이 공격 대상을 인식하는 표지판
ex)
코로나 바이러스 스파이크 단백질 전체가 항원이면,
그 중 항체가 실제로 달라붙는 부분이 에피토프
종류
- 선형 epitope: 아미노산이 일렬로 있는 서열 그대로 인식됨
- 구조적 epitope: 단백질이 접힌 상태의 3차원 구조로 인식됨
🖐 TCR (T-Cell Receptor, T세포 수용체)
T세포 표면에 있는 수용체 단백질
epitope(항원 조각)를 인식해서 면역 반응 유도
🔎 작동 원리
바이러스나 암세포의 항원 조각이 MHC 분자에 의해 제시됨
TCR이 이를 인식 → T세포가 활성화되어 공격 시작
🖐 MHC (Major Histocompatibility Complex, 주조직적합복합체)
세포 표면에 존재하면서, 세포 안에서 만들어진 항원 조각(peptide)을 T세포에게 보여주는 단백질 복합체
T세포에게 보여주는 신분증/신고서 ->
"내 안에 이런 단백질이 있어. 이상한 게 있으면 공격해 줘!"
역할
세포 안 단백질(정상 or 비정상) 일부를 절단해서 조각(펩타이드)으로 만듦 ->
이 조각을 MHC 단백질이 붙잡고 세포 표면으로 이동 ->
T세포 수용체(TCR)는 이 MHC+펩타이드 복합체를 스캔하고 ->
정상이면 → 무시
비정상(예: 바이러스, 암)이면 → 면역 반응 활성화
🖐 BCR (B-Cell Receptor, B세포 수용체)
B세포 표면에 있는 항체와 유사한 수용체
항원을 직접 인식 → 인식하면 항체 분비를 유도
🔎 차이점
TCR은 항원 조각을 MHC를 통해 간접적으로 인식
BCR은 항원 단백질 그 자체를 직접 인식 가능
🖐 클론(clone)
동일한 TCR/BCR을 가진 T세포 또는 B세포의 집단
한 개의 T세포/B세포가 특정 에피토프를 인식하고 활성화되면,
그 세포는 복제(clonal expansion)되어 수백만 개의 동일한 클론을 생성함
->
어떤 클론이 많은지 → 질병 유무나 면역 반응 여부를 판단하는 데 중요
🖐 바이오마커 (Biomarker)
질병의 존재, 진행 정도, 치료 반응 여부 등을 나타내는 생물학적 지표
✅ U-Map(Uniform Manifold Approximation and Projection) 이란?
고차원 데이터를 2차원 또는 3차원으로 시각화하기 위한 차원 축소 기법
- 데이터 간의 국소 구조(local structure)와 전체 구조(global structure)를 모두 잘 보존
- 클러스터링 구조를 시각화할 때 유용
-> 비슷한 발현 패턴을 가진 세포끼리 모여서 군집이 형성
🖐 예시
Binding 특성이나 single cell 데이터를 UMAP으로 2D에 투영하면,
환자군과 비환자군이 자연스럽게 분리되는지 시각적으로 확인 가능
->
시각적으로 "이 구역은 B세포, 저 구역은 NK세포"
이런식으로 클러스터를 확인 가능
✅ Singel cell 이란?
한 개의 세포 수준에서 생물학적 데이터를 측정하고 분석하는 기술
기존에는 수천~수백만 개 세포를 한꺼번에 분석했지만,
세포마다 유전자 발현이 다르다는 점에 주목해서 개별 세포의 특징을 파악하는 게 목적
1️⃣ 목적
세포 이질성 파악 (같은 조직 내에서도 세포별로 다름)
질병 상태에 따라 특정 세포 하위집단의 변화 감지
2️⃣ 왜 중요할까?
| 기존 Bulk 분석 | Single Cell 분석 |
|---|---|
| 여러 세포를 평균내어 분석 | 각 세포 하나하나의 발현을 분석 |
| 개별 세포의 특이성 파악 어려움 | 세포 이질성, 희귀 세포 포착 가능 |
| 병변이 섞여 있으면 구별 어려움 | 세포 하위 군집(클러스터) 구분 가능 |
ex)
암 조직을 bulk로 분석하면 모든 세포의 평균적인 유전자 발현만 나옴
→ 하지만 Single Cell은 암세포, 면역세포, 정상세포를 구분해서 분석 가능
3️⃣ 분석 절차
Step 1: 샘플 분리
혈액이나 조직에서 세포 하나하나를 분리
Step 2: mRNA 추출 및 시퀀싱 (scRNA-seq) -> 데이터를 만드는 과정
각 세포에서 mRNA 추출 → cDNA로 변환 → Sequencing
-
mRNA 추출
각 세포 안에 있는 mRNA(메신저 RNA)를 추출
이 mRNA는 세포가 지금 어떤 유전자를 얼마나 발현 중인지 보여주는 자료 -
cDNA로 변환
mRNA는 불안정해서 그대로 시퀀싱하기 어려움
-> 효소(reverse transcriptase)를 이용해 cDNA(상보적 DNA)로 바꿔줌
→ 이게 바로 유전자 발현 정보의 안정한 복사본 -
Sequencing (시퀀싱)
cDNA를 잘게 쪼개서 차세대 염기서열 분석(NGS)으로 읽음
결과적으로 각 세포마다 어떤 유전자의 조각이 몇 개나 있는지 파악함
Step 3: Preprocessing -> 전처리 과정
유전자 발현 행렬(matrix) 생성 (세포 × 유전자)
-
Normalization (정규화, 보정)
각 세포마다 전체 읽힌 유전자 수(read depth)가 다름
→ 이를 비교 가능하게 맞춰주는 작업이 필요함 -
Filtering (잡음 제거)
scRNA-seq 데이터는 노이즈(noise)가 많음
→ 필요 없는 세포나 유전자 제거함
Step 4: 차원 축소 (UMAP, t-SNE, PCA 등)
수천 개 유전자 정보를 2D 또는 3D로 축소
-> 유사한 세포끼리 모이게 시각화
Step 5: 클러스터링
비슷한 유전자 발현 패턴을 가진 세포끼리 묶음
-> 각 클러스터가 특정 세포 유형을 의미할 수 있음 (T세포, B세포, 암세포 등)
4️⃣ 어떤 정보들을 알 수 있는가
| 분석 결과 | 설명 |
|---|---|
| 세포 하위군(cluster) | 동일한 조직 내에도 여러 세포 종류가 섞여있음을 확인 |
| 특정 유전자 발현 | 각 세포군의 대표 유전자 파악 (marker gene) |
| 희귀 세포 탐색 | 전체의 0.1%도 안 되는 특이세포도 분석 가능 |
| 세포 간 전이 경로 | 세포가 어떤 경로로 분화/변화하는지 추정 (pseudotime analysis 등) |
5️⃣ 응용 예시
-
면역세포 분석 (ex. 감염, 암, 백신 연구)
T세포, B세포, NK세포 분리
-> TCR/BCR 다양성, 활성화 여부 분석(어떤 세포가 가장 중요한지 등) -
암 조직 분석
종양세포 vs 면역세포 vs 주변세포 구분
-> 종양 내 이질성(tumor heterogeneity) 분석 -
신경세포 분석
뇌에서 특정 뉴런 종류 탐색
-> 알츠하이머, 파킨슨병 관련 연구
🖐 종양 내 이질성(Tumor Heterogeneity)
같은 환자의 같은 종양 안에도, 다양한 성격의 암세포들이 섞여 있는 것(유전적, 기능적으로 다양한 세포 집단이 존재)
과거에는 종양 조직 전체를 한 번에 분석했기 때문에 평균값만 알 수 있었으나,
Single-cell 분석을 통해
- 개별 암세포 단위로 분석 가능
- 어떤 클론이 어떤 유전자를 발현하고 있는지 파악
🖐 왜 중요한가?
| 이유 | 설명 |
|---|---|
| 🎯 치료 저항성 | 항암제가 듣지 않는 세포가 일부 남아 재발 유발 |
| 🧬 정밀의료 | 모든 세포를 한 가지 약으로 죽이긴 어려움 → 맞춤 치료 필요 |
| 🧪 바이오마커 오차 | 어떤 세포는 발현, 어떤 세포는 X → 평균만 보면 잘못 판단 가능 |
ex)
환자의 폐암 조직 분석
70%의 암세포는 EGFR 돌연변이 → EGFR 억제제 사용
나머지 30%는 KRAS 돌연변이 → EGFR 억제제 안 듣고 재발
→ 이질성을 모르고 치료하면 반쪽짜리 효과
💡 4. Immcantation 보고 오기
-> Beacon 100개 단위로 나오는데 이 중 후보군 정하기
-> 그 후, mouse의 Singel cell 시각화
✅ Immcantation 이란?
면역 시스템 레파토리(repertoire) 의 sequencing 식 데이터 분석을 위한 파이프라인 모음
주로 항체, BCR(항체 서열), TCR 서열 데이터를 다룰 때 사용
1️⃣ 목적:
어떤 항체들이 생성되고 있는지 알기
특정 질병에 대한 면역 반응이 어떻게 일어났는지 분석
백신, 항암 치료, 자가면역 질환 연구 등
🔎
ex1) 코로나 백신 접종 후 → 어떤 BCR/TCR이 늘어났는지 분석 → 면역 반응 확인ex2) 암 환자 치료 전/후 → repertoire 비교
2️⃣ 왜 Immcantation이 필요할까?
방대한 면역세포 서열 데이터를 분석하려면
BCR/TCR을 자동으로 정렬하고 ->
클론 집단을 나누고 ->
돌연변이, 계통수 분석을 해야함
Immcantation은 이 과정을 자동화해주는 파이프라인
-> B세포/항체 데이터를 처음부터 끝까지 분석해주는 통합 도구 세트
🖐 면역 시스템 repertoire
우리 몸 안의 B세포, T세포가 인식할 수 있는 항원의 종류 총합
(얼마나 다양한 항원을 인식할 수 있느냐)
->
우리 몸이 만들어낼 수 있는 항체(Ab)나 BCR, TCR(T-cell receptor)의 다양한 서열 세트
(그 다양한 인식을 하는, BCR/TCR의 서열 다양성)
= 면역 repertoire
B세포 1: BCR 서열 A → 항원 A 인식
B세포 2: BCR 서열 B → 항원 B 인식
→ 이렇게 수많은 BCR/TCR 서열의 조합이 repertoire
🖐 항체 vs. BCR
| 구분 | 항체 (Antibody) | BCR (B cell receptor) |
|---|---|---|
| 정의 | B세포가 분비하는 단백질 | B세포 표면에 붙어 있는 수용체 |
| 위치 | 세포 밖 (혈액, 조직 등) | B세포 표면 (막에 고정) |
| 형태 | 분비형 | 막결합형 (membrane-bound) |
| 기능 | 항원을 중화하거나 면역작용 유도 | 항원을 인식해서 B세포를 활성화시킴 |
| 구조 | Ig 도메인 구조 (IgG, IgM 등) | 항체 구조 + 막 관통 부위 (transmembrane domain) |
BCR = B세포 표면에 붙은 항체 -> 세포에 붙어서 센서 역할
항체 = B세포가 항원을 만나 활성화된 후, 밖으로 분비한 BCR -> 세포 밖으로 나가서 공격 역할
🖐 BCR, TCR 서열 -> 항원 인식 도구
| 구분 | 설명 | 예시 |
|---|---|---|
| BCR (B Cell Receptor) | B세포 표면에 있는 수용체 (항체와 유사) | 감기 바이러스 항원과 결합하는 BCR |
| TCR (T Cell Receptor) | T세포 표면의 수용체. 항원을 MHC와 함께 인식 | 암세포 MHC와 결합된 항원을 인식하는 TCR |
인식 능력은 서열(sequence)에 따라 결정
-> 서열이 다르면 → 구조가 달라지고 → 인식 가능한 항원도 달라짐
🖐 면역 Repertoire 시퀀싱이란?
한 사람의 몸 안에 존재하는 수많은 B세포 / T세포의 BCR/TCR 서열을 한꺼번에 시퀀싱(염기서열 분석)하는 기술
✅ Immcantation 의 기능에는 무엇이 있는가?
Immcantation은 여러 개의 모듈/패키지로 구성되어 있음
전체적으로는 BCR/TCR 시퀀싱 데이터 → 분석 → 시각화까지의 흐름
| 모듈 | 주요 기능 | 요약 | 설명 |
|---|---|---|---|
| pRESTO | NGS 리드 품질 제어, UMI 처리 | 시퀀싱 데이터 전처리기 | FASTQ 파일 같은 원시 시퀀싱 데이터를 다듬는 역할. 품질 나쁜 리드 제거하고, UMI(Unique Molecular Identifier)를 활용해 PCR 중복 제거 등 수행. |
| Change-O | V(D)J 재조합 결과 처리, 클론 할당 | 서열을 그룹으로 나누기 | V(D)J 유전자 할당 결과를 바탕으로 유사한 BCR/TCR 서열들을 클론 그룹(clone)으로 분류 |
| SHazaM | SHM 분석, 거리 기반 클론 분류 | 돌연변이 분석 + 클론 분류 기준 계산 | 각 서열 간 얼마나 변이(SHM)가 일어났는지 분석 서열 거리 계산해서 같은 클론인지 판단하는 기준을 설정해줌 |
| TIgGER | 개별 유전자 할당 정확도 향상 | V(D)J 유전자 정확도 향상 | 서열이 너무 다양해서 기존 유전자 DB로 잘 맞지 않을 때, 샘플 안에서 새로운 유전자를 추정해줌 맞춤형 유전자 할당 도와주는 역할 |
| Alakazam | 시각화, 통계분석, 계통수 작성 등 | 시각화 도구 | 분석된 데이터를 기반으로 클론 분포, SHM 비율, 계통수(lineage tree) 등의 시각화 및 통계 처리 |
| Dowser | Single-cell 데이터 시각화 | Single-cell 시각화 도구 | 단일세포 분석 (예: scRNA-seq 데이터와 BCR/TCR을 연결해서 UMAP 같은 시각화)를 수행 |
🖐 NGS (Next-Generation Sequencing)
차세대 염기서열 분석 기술
유전자(DNA/RNA)를 대량으로, 빠르게, 저렴하게 읽는 기술
예전에는 하나하나 읽었지만, NGS는 수백만 개의 서열을 동시에 분석 가능
->
면역 repertoire 시퀀싱에서도 BCR/TCR 서열을 NGS로 얻음
🖐 UMI (Unique Molecular Identifier)
서열 하나하나에 붙이는 일련번호
PCR 복제 과정에서 중복이 생김 → 실제로 몇 개였는지 헷갈림
이를 막기 위해, 서열마다 UMI 태그를 붙임
->
BCR 서열의 중복 제거, 정확한 클론 수 추정에 중요.
🖐 PCR (Polymerase Chain Reaction)
DNA를 증폭시키는 기술
아주 소량의 DNA도 수백만 배로 복제할 수 있음
시퀀싱 전에 서열을 충분히 복제해서 읽기 쉽게 만듦
단, 중복이 생겨서 UMI 같은 보정 방법이 필요함
🖐 V(D)J 재조합
항체와 TCR의 맞춤형 조립 방식
BCR과 TCR은 고정된 게 아니라, V, D, J 유전자를 무작위로 조합해서 만들어짐
V = Variable
D = Diversity (BCR에만 있음)
J = Joining
이 조합 방식 때문에, 수백만 개의 다른 서열 → 수많은 항원 인식 가능
->
이걸 분석해서 "이 서열은 어떤 유전자 조합으로 만들어졌나?" 파악하는 게 IgBLAST, TIgGER 같은 도구의 역할
🖐 SHM (Somatic Hypermutation)
B세포가 스스로 돌연변이를 만들어내는 현상
항원에 반응한 후, B세포는 자신의 BCR 서열을 일부러 변이시킴
→ 더 잘 맞는 항체로 진화시키는 과정
->
SHazaM에서 이 SHM을 분석함. “어느 부위에서 얼마나 돌연변이가 일어났는가?” 같은 분석 가능
🖐 계통수 (Lineage Tree)
항체 서열의 진화 과정을 나무처럼 표현한 그래프
하나의 B세포 클론이 여러 번 SHM을 겪으면서 가지를 뻗음
->
그 계통을 트리로 표현 → 항체 진화도
🖐 scRNA-seq (Single-cell RNA sequencing)
세포 하나하나의 유전자 발현을 읽는 기술
일반 RNA-seq은 전체 세포 평균
scRNA-seq은 각 세포마다 어떤 유전자가 얼마나 발현되었는지 알 수 있음
->
BCR/TCR 시퀀싱과 함께 쓰면, 특정 B세포가 어떤 기능을 하는지도 분석 가능
🖐 IgBLAST
서열이 어떤 V(D)J 유전자 조합인지 판별해주는 도구
NCBI에서 제공하는 툴
시퀀싱된 BCR/TCR 서열을 입력하면:
사용된 V, D, J 유전자 / CDR3 서열 / 변이 위치 등을 분석해줌
🔎 전체 흐름 예시
- 실험실에서 B세포 RNA 시퀀싱 → FASTQ 파일 생성
- pRESTO로 전처리 (불량 리드 제거, UMI 정리 등)
-> 원재료 손질 (시퀀싱된 서열 다듬기)
- IgBLAST로 V(D)J 유전자 맞춤 → 결과를 Change-O에 넘김
- Change-O에서 클론 분류 + SHazaM으로 돌연변이/거리 분석
->
비슷한 음식끼리 분류 (비슷한 서열을 클론 그룹으로 묶기) +
맛 차이 분석 (서열 간 거리 = 돌연변이 정도 계산)
- 클론 정보 → Alakazam으로 그래프 그림(통계와 시각화)
- Single-cell로 분석된 경우 → Dowser로 UMAP 같은 시각화 가능
-> 세포 하나하나 위치까지 그려주는 지도 만들기 (Single-cell UMAP)
- TIgGER -> 새로운 레시피 추정 (새로운 유전자 패턴 찾아주기)
✅ Immcantation 사용법 익히기
https://github.com/immcantation
https://immcantation.readthedocs.io/en/stable/
실제 해볼 때, gpt 써가면서 하면 될 듯
✅ BEACON 이란?
Berkeley Lights에서 개발한 single-cell 분석 플랫폼
주로 B세포를 대상으로 항체 발굴(Antibody Discovery), 세포 분리, 기능 평가 등에 사용
💡 특징
| 기능 | 설명 |
|---|---|
| Single-cell level 작업 | 한 개의 세포를 하나의 나노펜(nanopen)에 담아서 추적/분석 |
| 분비능 분석 | 세포가 만드는 항체나 단백질을 실시간으로 모니터링 |
| 고속 스크리닝 | 수천 개의 세포를 한 번에 처리하고 성능 좋은 후보를 선택 |
| 자동화 | 이미지 기반으로 세포 이동/분석을 자동으로 수행 |
🔎 사용 예시 (B세포 분석의 경우)
- Mouse 에서 B세포를 분리
- Beacon 기기로 단일 B세포를 나노펜에 넣음
- 각각의 B세포가 생성하는 항체를 분석
- 항체 생산량, 특이성, 결합능 등을 기반으로 Top 후보군 선정
- 이 후보들을 bulk sequencing, scRNA-seq으로 분석 → Immcantation과 연동
💡 5. NGS, FastQ
Sorting 하는 것(Binding 유무 차이, 서열 차이)
✅ NGS 란?
Next-Generation Sequencing (차세대 시퀀싱)
유전자(DNA/RNA)를 대량으로, 빠르게, 저렴하게 읽는 기술
예전에는 하나하나 읽었지만, NGS는 수백만 개의 서열을 동시에 분석 가능
✅ FastQ 란?
NGS 데이터를 저장하는 대표적인 파일 형식
하나의 리드에 대해
- @로 시작하는 시퀀스 ID -> 해당 서열에 대한 식별자
- 서열 정보 (A, T, G, C)
- +로 시작하는 라인 (옵션 ID 반복)
- 품질 점수
(ASCII 문자로 인코딩된, 서열의 각 염기에 대한 base call quality)
4줄씩 반복되는 구조
@HISEQ:101:C6TKDACXX:4:1101:10000:100000
-> (서열 생성 장비 이름, 런 번호, 위치 정보 등)
GATTTGGGGTTT...
.+ -> 다음 줄이 품질 점수라는 걸 알려주는 구분자 역할
!''*((((***+...
✅ Sorting (Binding 유무 / 서열 차이)
BCR/TCR, 항체 등 서열 분석 후
binding 여부에 따라 분류하거나, 서열의 변이에 따라 정렬 가능
같은 V gene이지만 서열 차이로 binding 차이를 보이는 경우 → 이를 정렬하여 downstream 분석
💡 6. Colab 사용
QLoRA로 차원 축소하여 사용
✅ Colab에 QLoRA로 차원 축소하여 사용하는 것 익히기
1️⃣ 예시: ESM 모델 전체를 QLoRA 방식으로 Fine-tunning
2️⃣ 목적: 특정 task (예: binding 예측, classification 등)을 위해 ESM 같은 대형 PLM을 Colab에서도 학습 가능한 수준으로 저메모리 Fine-tunning
3️⃣ 방법:
ESM을 4bit로 양자화해서 메모리 아끼고
attention이나 FFN에 LoRA 모듈만 학습 가능하도록 추가
Colab에서도 큰 모델을 task에 맞춰 finetune 가능
🖐 “새로운 모델”에 QLoRA를 적용할 수 있는가?
안 된다, 또한 쓸 이유가 없다
QLoRA는 이렇게 정의됨:
“기존 사전학습된 대형 모델에 대해, 전체를 4bit 양자화하고 일부 모듈에 Low-Rank Adapter만 학습하여 효율적인 Fine-tunning 을 가능하게 하는 기술.”
🔻 즉, QLoRA는 다음 조건이 필수:
- 미리 훈련된 모델이 존재해야 함
- 그 모델 내부의 일부 layer (보통 attention/query/value/FFN 등)에 LoRA 모듈만 덧붙임
- 전체를 학습하지 않음 → 저비용
🛑 그런데 “새로운 Transformer를 학습”하는 경우엔?
모델이 사전학습되지 않았기 때문에, LoRA adapter를 붙인다고 성능 향상/절약은 의미가 없음
어차피 scratch 학습이면 모든 파라미터가 학습 대상 → QLoRA의 장점 (freeze + low-rank update)이 사라짐
결론: 새로 만드는 모델에는 QLoRA를 쓰지 않는다
🖐 Colab은 최대 24시간 제한인데, 일주일짜리 학습은 어떻게 하나?
1️⃣ Checkpoints 저장 후 이어서 학습
HuggingFace Trainer 또는 PyTorch에서 주기적으로 save_checkpoint 하도록 설정
Colab 세션이 끊겨도 다음 세션에서 이어서 로드 가능
2️⃣ 로컬/랩 서버로 이전
장기 학습이 필요한 경우엔 Colab보다 AWS, GCP, 개인 서버를 쓰는 게 현실적
특히 ESM, ProtT5, AlphaFold처럼 수일 이상 걸리는 경우는 Colab으로 불가능에 가까움
3️⃣ Epoch 수를 줄이고 task-specific adaptation으로 바꿈
요즘은 Full Finetuning보다
PEFT (QLoRA 포함), Adapter Tuning
같은 경량화된 방법으로 성능 확보하면서 짧은 학습에 집중하는 경향
💡 7. 서버 GPU 1개, 4090
✅ GPU 4090 의 사양 파악
1️⃣ 4090으로 가능한 Task들
| Task | 가능 여부 |
|---|---|
| ESM2 650M 추론/임베딩 | 아주 쾌적함 |
| ESM2 650M QLoRA 파인튜닝 | Colab 기준도 충분히 가능 |
| BERT/T5 파인튜닝 (base) | 무난 |
| Protein structure prediction (AlphaFold-multimer) | 가능 (메모리 충분) |
| Diffusion model 훈련 | 가능하나 VRAM 24GB로 일부 제약 있음 |
| GPT-style 모델 훈련 (1B+) | Full fine-tuning은 무리, QLoRA는 가능 |
| Transformer 기반 CNN, MLP 등 자체 모델 훈련 | 매우 빠름 |
| Batch size ↑ (multi-GPU 수준 학습) | 메모리 제한으로 어려움 |
2️⃣ 현업용 GPU (A100, H100 등)과 비교
| 항목 | RTX 4090 | A100 (40GB) | H100 (80GB) |
|---|---|---|---|
| FP16 성능 | 66 TFLOPs | 312 TFLOPs | 989 TFLOPs |
| VRAM | 24GB | 40GB | 80GB |
| 메모리 대역폭 | 1008 GB/s | 1555 GB/s | 2039 GB/s |
| Multi-instance GPU | 미지원 | 지원 | 지원 |
| 가격 | 250~300만 원 | 2,000만 원 이상 | 4,000만 원 이상 |
| 목적 | 소비자/개인 연구자 | 연구소, 기업용 | 초대규모 AI 학습 |
💡 8. GroMax(MD)
✅ MD(Molecular Dynamics) 란?
분자들이 시간에 따라 어떻게 움직이는지를 시뮬레이션하는 계산 방법
원자 및 분자의 움직임을 뉴턴의 운동 법칙에 따라 시간에 따라 계산
목적:
단백질 구조의 안정성 평가, 결합 메커니즘 분석, 구조 기반 약물 설계 등
🔎 예시:
단백질이 수용액 속에서 어떤 식으로 접히는가?
리간드가 단백질에 어떻게 결합하는가?
->
초기 구조 (PDB) + force field → 시뮬레이션 진행
수 ns~μs 단위로 원자 움직임 추적
✅ Gromax 란?
GROningen MAchine for Chemical Simulations
빠르고 효율적인 MD 시뮬레이션 툴로, 주로 바이오분자(단백질, DNA 등)에 사용
-> 단순 단백질 구조 보는 프로그램이 아니라,
그 구조가 물리 법칙 아래에서 어떻게 움직이고 상호작용하는지를 보는 실험 대체 도구
✅ Gromax의 기능에는 무엇이 있는가?
| 기능 | 설명 |
|---|---|
| 에너지 최소화 | 시작 구조의 물리적 비현실성 제거 |
| MD 시뮬레이션 | 시간에 따른 분자의 움직임 시뮬레이션 |
| 온도/압력 조절 | NVT, NPT ensemble 설정 가능 |
| 제약 (Constraints) | 특정 결합이나 각을 고정하여 계산 안정화 |
| 분석 도구 제공 | RMSD, RMSF, 거리, 반응 좌표 등 다양한 분석 |
| 병렬처리 및 GPU 지원 | 대규모 계산에도 빠르게 실행 가능 |
✅ Gromax 사용법 익히기
http://www.mdtutorials.com/gmx/
🔎 전체 워크플로우 요약
1️⃣ pdb2gmx -f input.pdb -o processed.gro -water spce
: PDB 구조파일을 force field와 함께 .gro 형식으로 변환
2️⃣ editconf -f processed.gro -o boxed.gro -c -d 1.0 -bt cubic
: 시뮬레이션 박스 생성
3️⃣ solvate -cp boxed.gro -cs spc216.gro -o solvated.gro -p topol.top
: 물 분자 삽입 + (이온 추가 및 중성화)
4️⃣ grompp -f em.mdp -c solvated.gro -p topol.top -o em.tpr
mdrun -v -deffnm em
: 에너지 최소화 수행(초기 구조에서 비현실적인 충돌 제거)
5️⃣ grompp -f nvt.mdp -c em.gro -r em.gro -p topol.top -o nvt.tpr
mdrun -deffnm nvt
: 일정 온도 유지하는 NVT 시뮬레이션 수행
6️⃣ grompp -f npt.mdp -c nvt.gro -r nvt.gro -t nvt.cpt -p topol.top -o npt.tpr
mdrun -deffnm npt
: 일정 압력까지 유지하는 NPT 시뮬레이션 수행
7️⃣ grompp -f md.mdp -c npt.gro -t npt.cpt -p topol.top -o md.tpr
mdrun -deffnm md
: 실제 시뮬레이션 실행 (실제로 분자들이 시간에 따라 어떻게 움직이는지 시뮬레이션)
8️⃣ 추가 팁
.mdp 파일: 각 단계의 설정 (시간, 온도, 압력, 시뮬레이션 옵션 등)을 포함
.gro, .top, .tpr, .xtc 등 다양한 형식의 입출력 파일 이해 필요
시뮬레이션 결과 분석 단계에서는 단백질의 안정성(RMSD), 유연성(RMSF), 반지름, 결합 거리, 수소결합 수, 자유 에너지 계산 등 분석
✅ 어디에 쓸 수 있나?
GROMACS는 실세계에서 분자들이 어떻게 움직이고 상호작용하는지를 물리 기반으로 시뮬레이션해 주는 툴
->
이걸 통해 “그냥 구조만 보는 것”이 아니라, 구조의 행동을 분석할 수 있기 때문에 실제 응용으로 이어질 수 있음
1️⃣ 약물 설계 (Drug Design)
리간드가 단백질 포켓에 얼마나 잘 결합하는지 확인
단백질과 약물(리간드)을 복합체 형태로 시뮬레이션 →
시간이 지남에 따라 리간드가 안정적으로 결합을 유지하는지 관찰
리간드가 binding pocket에서 튀어나가거나, 움직임 없이 고정되어 있는지 분석
📊 분석 도구:
gmx distance: 결합 거리 분석
gmx hbond: 수소결합 유지 여부 확인
gmx energy: 결합 에너지 추정
이를 통해 결합 친화도 추정 → docking보다 더 현실적인 결합 평가 가능
2️⃣ 단백질 구조 연구 (Protein Stability & Dynamics)
단백질이 접힌 상태에서 안정적인가?
구조가 어떻게 흔들리는지, 펼쳐지는지 분석
구조를 그대로 넣고 MD 돌리면 구조가 자연스러운 물리 환경에서 안정한지 확인 가능
ex) 어떤 돌연변이를 넣었을 때 단백질이 불안정해져서 흐트러지는지 확인
📊 분석 도구:
gmx rms: 전체 구조의 RMSD 변화 (안정성)
gmx rmsf: 개별 잔기의 유연성 (loop의 흔들림 등)
gmx gyrate: 구조 압축도 (compactness)
3️⃣ 막 단백질 연구 (Membrane Proteins)
지질 막 속에서 단백질이 어떻게 움직이고, 각도 변화가 있는지, 채널을 여닫는지 관찰
GROMACS는 막 단백질 + 지질 이중층 (lipid bilayer) 시뮬레이션 가능
막 환경에서의 삽입 안정성, 방향성, 지질과 상호작용 분석 가능
ex) 이온 채널이 막에서 열릴 때 구조가 어떻게 변하나?
4️⃣ 백신 개발 (항원-항체 상호작용)
항원과 항체가 결합한 복합체의 안정성과 결합 방식을 시간에 따라 관찰
항체의 paratope와 항원의 epitope 사이의 결합 유지 여부, 수소결합 유지, 접촉면 변화를 분석
백신 후보 항원을 설계한 후, 항체와 결합이 잘 유지되는지 시뮬레이션으로 미리 확인