본 포스트에서는 scRNA-seq의 raw data preprocessing에서 일어날 수 있는 이슈들을 설명한다.
scRNA-seq의 시퀀서는 Binary Base Cell (BCL) 파일을 생성한다.
BCL 파일은 시퀀싱 output을 바이너리 형태로 저장하고 있기 때문에,
저장공간 차원에서는 장점이 있으나, 인간이 이해하기 힘들다는 단점이 있다.
따라서 BCL 파일은 text-based sequence file format인 FASTQ로 변환 후, 추가적인 분석을 진행한다.
문맥에 따라 raw data가 BCL format이거나 FASTQ format일 수 있지만,
여기서는 FASTQ format을 raw data로 지정한다.
Seurat이나 Scanpy를 통해 진행되는 scRNA-seq의 대부분의 분석들은 count matrix와 진행된다.
Count matrix는 row가 gene을, column이 cell을 나타내는 matrix로,
서로 다른 셀 내에서 각 유전자들의 발현 정보를 담고 있다.
따라서, Raw data preprocessing은 read의 서열 정보 만을 담고 있는 FASTQ 파일로,
세포와 유전자 정보까지 추가된 Count matrix를 생성하는 과정을 뜻한다.
Count matrix는 앞으로 진행되는 모든 분석의 input이 되기 때문에,
raw data processing에서 문제가 있을 경우 모든 분석과 추론이 무의미 해진다.
raw data processing 과정의 몇 가지 중요한 challenge들과
그를 위한 computational methodology들을 알아보자.
1. Row data quality control
FASTQ 파일을 생성한 후에는 FastQC 프로그램을 통해 read quality를 손쉽게 확인 할 수 있다.
FastQC는 input FASTQ 파일의 QC report를 생성한다.

QC report는 각 항목에 대해 통과, 실패, 경고 등을 정리하여 보여준다.
각 항목들을 살펴보자.
-
Basic statistics
Basic statistics는 파일 형식, 인코딩, 서열 수, FASTQ 내 poor quality 플래그 수, sequence length, GC 비율 등의 기본적인 통계 정보들이 담겨있다.
좋은 데이터의 경우, poor quality 서열이 적고, uniform한 sequence length를 보이며,
분석한 생물의 overall GC content를 보여야 한다. -
Per Base Sequence Quality
Sequence length 내 염기 서열 위치 별 quality 분포를 보여준다.
좋은 데이터의 경우, length 전반적으로 동일한 수준의 quality를 보여야 한다.
-
Per Tile Sequence Quality
각 flow cell tile 내 read 들의 평균 quality의 편자(deviation)을 보여준다.
flow cell tile은 Illumina 사의 시퀀서에서 사용되는 sequencing section이다.
좋은 데이터의 경우, 편차가 없이 동일해야 한다. -
Per Sequence Quality Scores
파일 내 read들의 평균 Quality Score의 histogram 분포를 보여준다.
좋은 데이터의 경우, 높은 평균에 분포가 집중되어 있어야 한다. -
Per Base Sequence Content
A,C, G, T 각 염기의 sequence length 내 분포를 보여준다.
시작 부분은 priming site이기 때문에 분포가 편향되어 있을 수 있지만,
좋은 데이터는 이외 부분에서 uniform한 분포를 보여야 한다. -
Per Sequence GC Content
read 들의 GC content 분포를 이론적인 분포와 비교하여 보여준다.
좋은 데이터는 분포의 peak 부분이 서로 일치해야 하고, 분포가 비슷해야 한다. -
Per Base N Content
Sequence length 내 N base의 분포를 보여준다.
N base는 ACGT 염기들 중 하나를 confidential 하게 할당 할 수 없을 경우 할당하는 염기이다.
좋은 데이터는 N base를 가지지 않아야 한다 -
Sequence Length Distribution
read들의 legnth 분포를 보여준다.
좋은 데이터는 모든 read들이 거의 같은 length를 보여야 한다. -
Sequence Duplication Levels
read sequence 들의 duplication 정도를 보여준다.
좋은 데이터의 경우 낮은 duplication level을 보여야 한다.
하지만, scRNA-seq에서 사용하는 UMI를 FastQC는 인식하지 못하기 때문에,
Highly expressed gene의 많은 read 수를 duplication으로 인식하여
warning을 띄우는 경우가 있으나, 이 경우 data quality의 문제는 아니다. -
Overrepresented Sequences
전체 sequence read 중 0.1% 이상 발현된 sequence의 count와 percentage를 보여준다.
역시나 highly expressed gene의 경우 과발현으로 인식되는 경우가 있다.
Possible Source 부분이 No hit이 아닐 경우, 해당 Source로 인한 library 오염을 의심할 수 있다. -
Adapter Content
염기 서열 내에서 관측된 adapter sequece의 누적 비율을 보여준다.
좋은 데이터의 과하게 관측되는 adapter가 존재하지 않아야 한다.
더 자세한 내용은 FastQC의 공식 설명을 참고하자.
https://www.bioinformatics.babraham.ac.uk/projects/fastqc/Help/3 Analysis Modules/
2. Alignment / Mapping
Alignment 혹은 Mapping은 sequence read의 potential genomic loci를 결정하는 과정이다.
즉, 유전체 내에서 어떤 부분에서 유래된 read인지를 맞추는 과정이다.
bulk RNA-seq부터 많은 method들이 연구되어 왔지만,
single cell sequencing에서는 사용이 힘든 이유가 몇 가지 있다.
- Cell barcode(CB)와 UMI의 위치나 길이가 platform 별로 다양하다.
- CB와 UMI가 sequecing aligment를 방해한다.
- 수 억, 수 십억의 세포를 다루기 때문에 계산에 메모리나 시간적 문제가 있다.
따라서 Cell Ranger와 같은 scRNA-seq 특화된 Alignment 도구가 개발되었다.
Alignment에 앞서 다양한 고려 사향이 있다.
- 어떤 Alignment algorithm을 사용할지
- spliced alignment, contiguous alignment, lightweight-mapping
- Global mapping or local alignment
- 어떤 reference를 사용할지
- Whole genome
- Transcriptome
- Augmented transcriptome including intron region
각 tool마다 위와 같은 전략이 다르기 때문에 자신의 데이터 셋과 목적에 맞게 선택해야 한다.
3. Cell barcode(CB) correction
Droplet-based single cell 분리 시스템에서는 각 세포가 barcoded bead와 함께 droplet에
encapsulated 된다. bead 들은 세포 안의 RNA content가 어떤 세포에서 유래된 것인지
표시하기 위한 unique한 염기서열 tag, Cell barcode(CB)를 포함한다.
droptelt-based library의 Barcoding 과정에서 일어날 수 있는 몇 가지 error를 알아보자.
- Doublet / Multiplet: 한 droplet 안에 여러 세포가 포함되어 동일한 CB가 중복 사용될 경우
- Empty Droplet: droplet이 세포를 담지 못하고, barcode만 잘못 인식되어 세포 수를 overcounting
- Sequence error: PCR이나 sequencing 과정에서의 요류
이러한 오류를 바로잡기 위한 몇 가지 대표적인 방법들이 개발 되었다.
-
Known list of potential barcode
platform 내에서 생성한 potential barcode list와 관측된 barcode를 대조한다.
barcode 자체가 잘못 sequencing 되는 경우가 존재하기 때문에
potential vs observed barcode 간의 hamming distance, edit distance 대조를 통해 오류를 수정한다.
문제점이 있다면 CB가 potential list 내에서 중복 확인 될 수 있다는 점이다.
-
Knee or elbow based method
외부 자료의 추가 없이, 관측된 barcode들만을 가지고 통계적으로 correction 하는 방법이다.
high-quality barcode는 많이 관측될 것이라는 가정과 함께,
누적 빈도가 낮은 barcode, elbow point 이하의 barcode를 제외시킨다.
문제점이 있다면, 확실한 elbow point가 안보일 수도 있고, 너무 보수적으로 판단 할 수 있다는 점이다.
-
Filtering based expected cell count
2번의 방식과 비슷하게 관측된 데이터 만을 가지고 correction 하는 방법이다.
사용자가 분석된 cell의 양을 예측하여 특정 percentile 이상의 barcode만 채택하는 것이다.
채택되지 않은 barcode들은 1번의 방식으로 수정 후 다시 검토가 가능하다.
4. UMI resolution
CB correction로 세포 차원 문제를 해결했다면, 다음은 세포 내 각 gene의 양을 측정해야 한다.
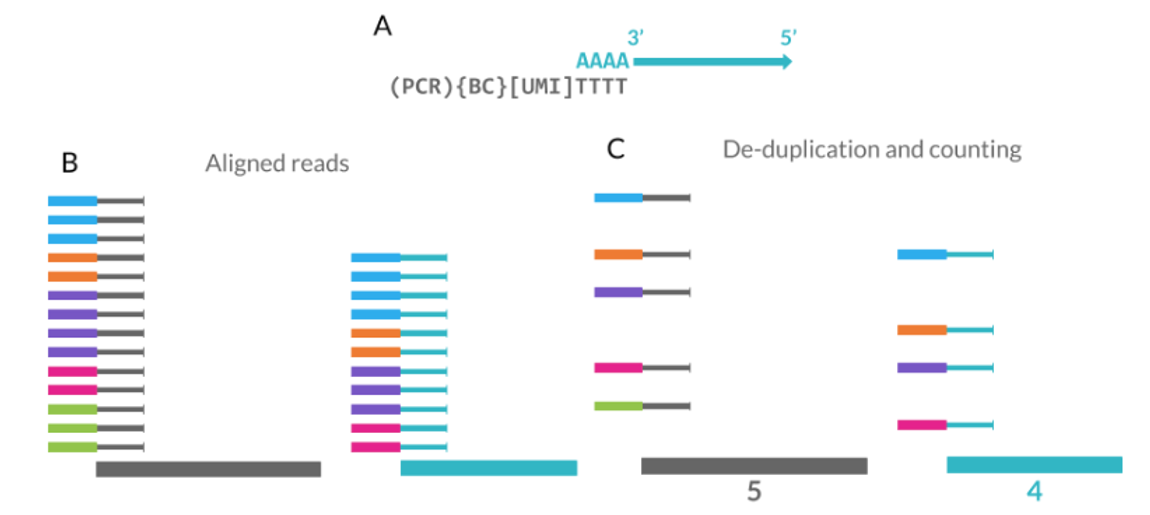
Unique molecular identifier(UMI)는 짧은 염기서열로 세포 내 molecule, RNA를 unique하게 tag한다.
Sequencer는 sequencing의 confidence를 높이고, 오류를 수정하기 위해 PCR 과정을 거친다.
이를 통해 동일한 molecule이 증폭되고, 중복된 sequencing이 이루어진다.
UMI는 amplification 이전에 부착되므로, De-duplication을 통해 실제 count를 관측할 수 있게 한다.
즉, 동일한 UMI를 가진 read들은 하나의 molecule에서 유래되었다고 생각하는 것으로,
amplification 과정에서 생길 수 있는 bias를 해결할 수 있다.
UMI resolution은 관측된 UMI와 연관된 read들을 processing하여, 각 유전자에서 유래된
original number of molucule, 전사체 발현량을 추측하는 과정이다.
하지만 UMI resolution에는 몇 가지 challenge가 존재한다.
- UMI sequecing 오류: PCR이나 시퀀싱 과정에서의 에러로 인식 불가
- UMI mapping 오류: UMI tagged RNA 서열이 중복 mapping 되는 경우
- Convergent: same UMI is used to different molecules from the same gene
- Divergent: two or more UMI from the same molecule
이러한 오류를 고려하며 UMI resolution을 하기 위한 대표적인 툴로는 UMI-tools가 있다.
Pypi, conda, github로 제공되며, UMI resolution과 count matrix 생성을 지원한다.
https://github.com/CGATOxford/UMI-tools
5. Count matrix quality control
count matrix를 생성 후에도 quality control이 중요하다.
count matirx QC를 위해 사용되는 측정 항목들로는 다음이 있다.
- Total mapped reads
- Distrubution of UMI per cell
- Distribution of UMI deduplication rate
- Distribution of detected genes per cell
- Mitochondrial gene expression percentage
위와 같은 사항들을 고려하여 empty droplet이나 multiplet을 filtering 한다.
앞서 언급된 Knee 와 Elbow point를 활용하여 필터링 하는 것도 가능하고,
이를 위한 여러가지 통계적 방법 또한 연구되고 있다.
Count matrix를 생성 하기 까지, 여러 heuristic이나 filtering이 적용되었다.
이는 raw sequencing으로부터 정보 소실이 있었다는 것을 의미하기 때문에,
count matrix에 raw data의 정보를 이식하는 방법 또한 연구되고 있다.
위의 preprocessing 과정은 terminal에서 alevin-fry 혹은 simpleaf를 통해 진행할 수 있다.
