tata box란?
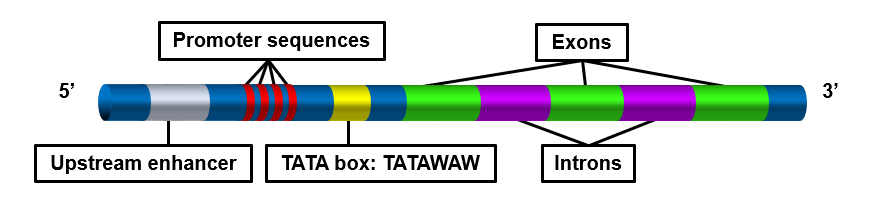
유전적 시퀀스가 읽혀지고 디코드 되는 곳을 나타내는 DNA sequence.
promoter sequence의 타입으로 전사(transcription)이 시작되는 다른 분자를 지정함.
(출처: https://www.nature.com/scitable/definition/tata-box-313/)
Alphabet 모듈
tatabox_seq = Seq("tataaaggcAATATGCAGTAG", IUPAC.unambiguous_dna)
print(tatabox_seq)책에서는 이렇게 코드 작성하면 알파벳 모듈을 사용할 수 있다고 되어있는데 에러 발생함
-> Bio.Alphabet has been removed from Biopython.
결론은 바이오파이썬에서 alphabet 모듈을 삭제함.
Bio.Alphabet의 두 가지 메인 기능에는
1) Record the molecule type (DNA, RNA or protein) of a sequence
2) Declare the expected characters in a sequence, alignment, motif, etc.
가 있는데 알파벳 객체의 의도된 목적이 정확하게 정의되지 않았고 오래 되기도 했고 복잡했다는 이유로 삭제하기로 했다고 함.
**old style**
from Bio.Seq import seq
from Bio.Alphabet import IUPAC
tatabox_seq = Seq("tataaa", IUPAC.unambiguaous_dna)
print(tatabox_seq)⬇️
**new style**
from Bio.Seq import seq
tatabox_seq = Seq("tataaa")
print(tatabox_seq)Sequence 객체 다루기
Sequence 객체 만들기
tatabox_seq = Seq("tataaaggcAATATGCAGTAG")
print(tatabox_seq) #tataaaggcAATATGCAGTAG
print(type(tatabox_seq))count 서열 세기
.count(" ")
ex) print(exon_seq.count("A")) #3GC-contents(%) 계산하기
#전체 서열 중 G or C의 염기를 세어 전체 서열 중 G,C염기 함량이 얼마나 되는지 구함
exon_seq = Seq("ATGCAGTAG")
g_count = exon_seq.count("G")
c_count = exon_seq.count("C")
gc_contents = (g_count + c_count) / len(exon_seq)*100
print(gc_contents) #44.44서열 대소문자 변형
.upper()
.lower()
ex) print(tatabox_seq.upper()) #TATAAAGGCAATATGCAGTTAG
ex) print(tatabox_seq.lower()) #tataaaggcaatatgcagttag서열 객체 transcribe(전사), translate(번역)
dna = Seq("ATGCAGTAG")
mrna = dna.transcribe() #전사
ptn = dna.translate() #번역
print(mrna) #AUGCAGUAG
print(ptn) #MQ*
여기서 * 기호는 단백질 번역 과정이 끝났음을 알리는 종결 코돈
3s as Semin Sarah Serika