https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8545213/
논문 리뷰에 앞서
논문 리뷰는 다회독을 하며 내용을 수정해 나갈 게시글입니다.
현재 1회독 중이며, Abstract, Introduction, Conclusion을 정리합니다.(22.09.28)
현재 2회독 중이며, 세부 내용을 정리합니다. (22.10.02)
6. Result를 제외한 내용 리뷰 완료 (22.10.25)
1회차 리뷰
Abstract
해당 논문에서는 Genorm Seq에 Regulatory Motif가 포함되어있는지를 확인합니다.또한, SARS COV-2를 다른 코로나 바이러스와 식별하기 위해 CNN-Bi-LSTM 모델을 제안하고 있습니다.
Introduction
SARS-CoV-2 바이러스와 다른 코로나 바이러스들은 유전적 유사성 때문에 서로를 구별하기가 어렵습니다.
현재는 RT-qPCR을 이용해 환자의 SARS-CoV-2 감염을 확인합니다.
RT-qPCR은 RT-PCR과 qPCR을 결합하여 qPCR 반응결과에서 cDNA를 사용하여 RNA level을 측정합니다.
관련 논문 2 논문에 따르면, RT-qPCR은 ORF1ab와 N유전자를 이용하여 SARS-CoV-2를 식별합니다.
그러나, RT-qPCR을 이용한 검사에서 실제 감염된 환자들에게서 잘못된 음성 분석이 확인되었습니다.
해당 논문에선 이를 바이러스의 RNA서열에서의 변이가 이유가 될 수 있다고 보았습니다.
따라서 해당 논문에선 SARS-CoV-2와 다른 코로나 바이러스를 식별하기 위해 새로운 방법들을 탐구합니다.
regulatory motif
motif는 유사한 sequence 그룹에서 반복적으로 발생하는 짧은 근사 nucleotide sequence입니다.
regulatory motif는 유전자의 발현을 조절하는데 사용됩니다.
Transcription factors(TFs)는 DNA에 붙는 단백질로, DNA를 RNA로 변환하거나, 전사하는 역할을 합니다.
TFs는 유전자 발현과정에서 DNA Seq와 결합하고, 유전자의 발현여부를 결정하게 됩니다.
해당 논문에서는 주어진 Seq에 SARS-CoV-2를 발현시키는 regulatory motif가 포함되어있는지 확인하는 과정에서 SARS-CoV-2끼리 공유하는 regulatory motif에 주목합니다.
이를 위해 SARS-CoV-2의 regulatory motif가 포함되어 있는지 확인하는데 중요한 nucleotide의 탐지에 중점을 둡니다.
또한, 이들은 최신 CNN-Bi-LSTM을 통합해 regulatory motif를 포함하며, SARS-CoV-2와 다른 코로나 바이러스를 분류하는 하이브리드 딥러닝 알고리즘을 제안합니다.
Problem Statement
주어진 Seq에서 SARS-CoV-2의 regulatory motif가 포함되어있는지 확인하고, SARS-CoV-2를 다른 코로나 바이러스들 사이에서 식별하는것은 Binary Classification 문제로 볼 수 있습니다.
Conclusion
SARS-CoV-2 바이러스와 다른 비슷한 바이러스를 비교하기 위해서, 주어진 DNA Seq가 SARS-CoV-2의 것인지 아닌지 식별하는 것은 매우 중요합니다.
해당 논문에서 제안된 CNN-Bi-LSTM 모델은 주어진 Seq에 SARS-CoV-2의 regular motif가 포함되어 있는지 확인 할 뿐 아니라, SARS-CoV-2를 다른 코로나 바이러스와 식별하는 데 성공했습니다.
또한, saliency map을 이용해 주어진 DNA Seq에서 SARS-CoV-2의 regulatory motif를 식별하는 데 중요한 역할을 하는 염기 및 nucleotide를 식별 할 수 잇었습니다.
2회차 리뷰
Related Work
현재 사용되고 있는 genorm seq 분석 기술(BLAST)는 정렬을 필요로 합니다.
그러나, 이런 기술은 많은 계산시간이 필요합니다.
또한, 이러한 정렬을 이용한 기술들은 유전체가 homologous하며, 동일한 연속 구조를 가진다고 가정하지만, 실제로는 항상 이런 가정이 설립하지는 않습니다.
때문에 유전자의 정렬이 필요하지 않은 새로운 방법이 연구되었고, feedforward CNN을 사용한 Deepfam와 MLDAP-GUI가 제안되었습니다.
해당 연구에선 DNA의 시간적 효과(temporal effects)를 포착하는 CNN층과 Bi-LSTM층으로 구성된 CNN-Bi-LSTM모델을 제안했습니다.
DNA genorm seq에는 A, T, C, G의 네가지 base가 있으며, 이 4개의 염기쌍은 염기쌍 {A, T, C, G}를 형성합니다.
해당 연구에선 이 염기쌍을 A, T, G, C 네가지의 문자 패턴으로 간주하고, One-hot Vector를 사용하여 텍스트 데이터와 유사한 방식으로 염기쌍을 표현합니다.
관련 논문 12
DNA seq를 단어로 변환하는 데 관련 논문 22과 같은 절차를 사용하였습니다.
(변환절차에 대한 자세한 내용은 관련 논문 22에 대한 Review, DNA Seq Classification by CNN 논문 에서 설명하였습니다.)
관련 논문 30에서는 SARS-CoV-2를 다른 코로나 바이러스 사이에서 분류하고, SARS-CoV-2의 regulatory motif를 식별하기 위해 DeapSEA와 유사한 DanQ를 개발했습니다.
해당 논문에서는 Deep Sea관련 논문 24의 모델에서 CNN과 Bi-LSTM의 순서를 바꾸었습니다.
Materials and Methods
해당 연구는 SARS-CoV-2 바이러스를 다른 코로나 바이러스와 분류하는 관련 논문 2의 연구를 확장합니다.
또한, DNA binding motif를 예측하기 위한 관련 논문 20의 연구를 확장합니다.
두 연구를 확장하기 위해 CNN-Bi-LSTM을 사용합니다.
Datasets
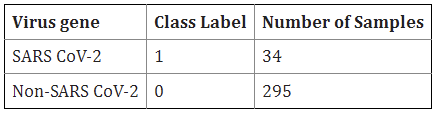
해당 연구에 사용된 329개의 샘플은 모두 코로나 바이러스에 속하며, SARS-CoV-2의 샘플은 1, 나머지 샘플은 0의 클래스를 가집니다.
해당 데이터셋은 positive class인 SARS-CoV-2가 10.3%로 불균형을 이루고 있습니다.
Algorithms
이번 장에서는 해당 연구에서 제안된 모델 CNN-Bi-LSTM이 사용 한 각각의 모델들을 설명합니다.
CNN
일반적으로 Text data를 2차원 배열로 변환하기 위한 단위는 단어 입니다.
하지만 DNA Seq 데이터는 A, T, G, C의 네가지 글자로 이루어진 text data 입니다.
이 데이터는 일반 text data와 달리 단어 사이에 빈 공간이 없기에 단어라는 단위를 이용하여 데이터를 변환할 수 없습니다.
때문에 Seq를 연속되는 세 자리씩 읽으면서 임의로 특정 단어를 만들어 냅니다.
이후 만들어진 단어들을 one-hot vector로 변환하여 CNN에 입력을 하게 됩니다.
참고 : DNA Seq Classification by CNN 논문
LSTM
LSTM은 colah의 블로그에서 매우 잘 설명하고 있습니다.
논문에서 사용한 그림에 누락된 부분이 있어 해당 블로그의 설명을 리뷰하는 글을 따로 작성해 LSTM 설명을 대체하겠습니다.
Bi-LSTM
예측을 하는 데 있어 선행 입력과 후속 입력을 모두 고려하여 예측값을 결정해야 하는 상황이 있습니다. 이러한 상황을 해결하기 위해 해당 연구에서는 Bi-LSTM을 제안합니다.
다음의 그림은 LSTM이 시간 에서 시간 로 진행되는 순방향으로의 네트워크를 계산하는 forward layer를 가지고 있음을 보여줍니다.
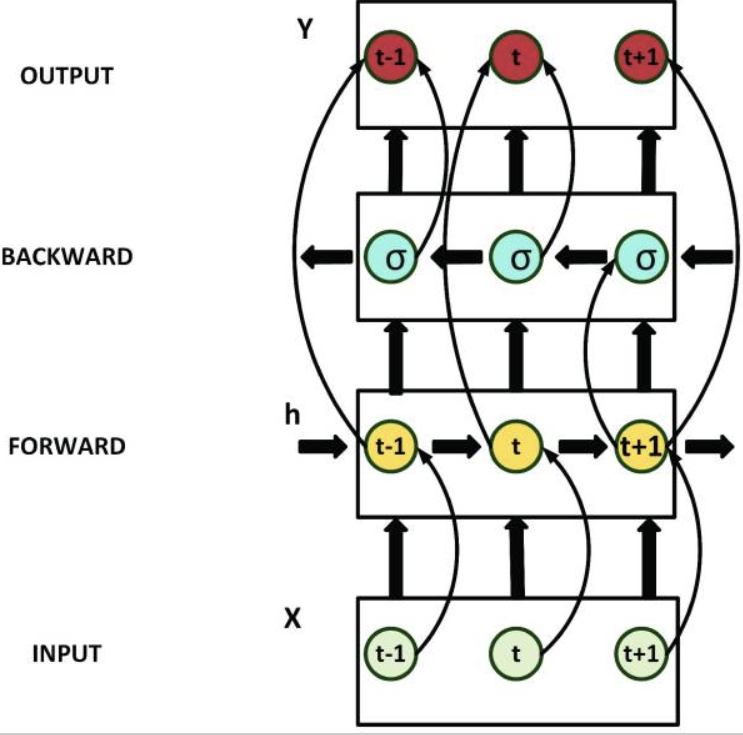
이렇게 계산된 forward layer의 출력값이 저장된 후 backward layer에서는 시간 에서 시간 으로 진행되는 역방향 네트워크 계산이 이루어지고, 저장 됩니다. 이렇게 저장 된 forward layer의 결과값과 backward layer의 결과값은 Concat (이어 붙이기), Mul(곱), Su(합), Ave(평균)의 네가지 방법 중 하나를 사용하여 처리됩니다.
Bi-LSTM을 사용하는 대부분의 연구에서 기본적으로 Concat의 방법을 사용하기에 이번 연구 역시 Concat의 방법을 사용하여 출력을 처리했습니다. 이 방법은 입력 벡터의 크기에 비해 출력 벡터의 크기를 두배 증가시키기 때문에 더 나은 성능과 더 낮은 log loss를 가져옵니다. 또한, vanishing/exploding gradient 문제를 해결하기 위해 시간을 기반으로 한 역전파 (BPTT)를 사용하여 모델을 훈련시켰습니다.
Proposed Architecture
아래 그림은 이 연구에서 사용 한 모델의 구조를 보여줍니다.
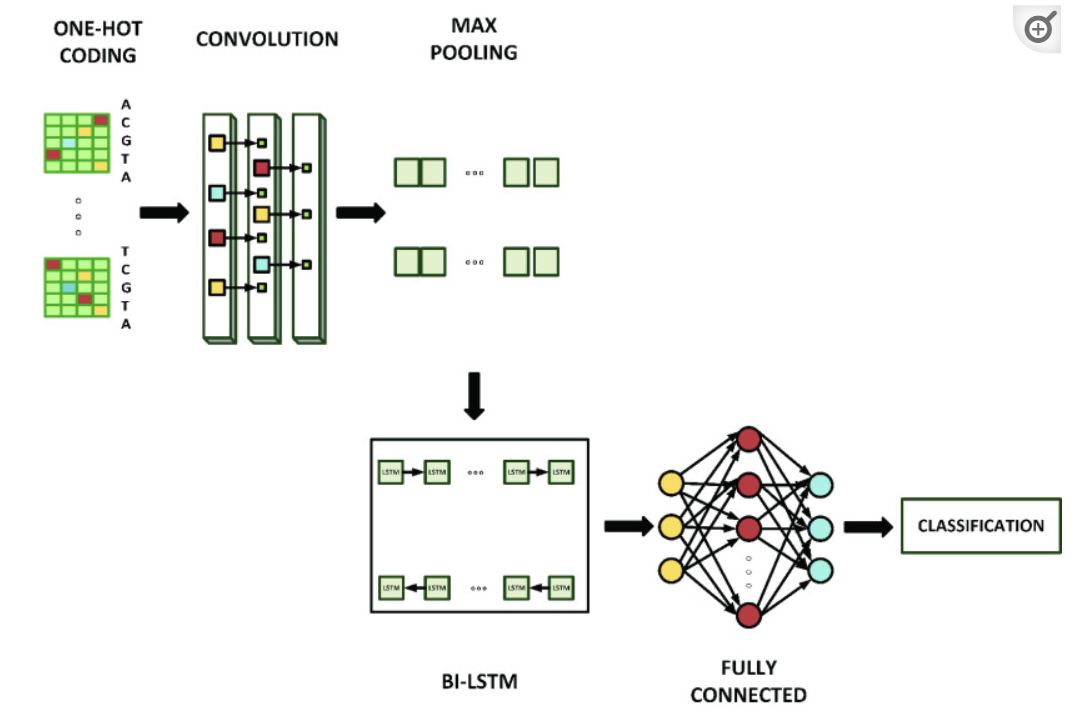
해당 연구에선 3층의 CNN과 Max-poolingdmf 사용하여 입력 행렬에서의 특징을 추출합니다.
이후 Bi-LSTM과 DENSE층이 차례로 연결되어 출력을 담당합니다.
해당 모델은 overfitting을 억제하기 위해 Dropout을 사용하였으며, 입력에서 고차원의 One-hot vector를 해 Coding/Encoding층과 word embedding층을 대체하였습니다.
Experiments
해당 연구에서는 데이터의 전처리로 각 게놈 서열이 SARS-CoV-2의 서열(Positive sample)인지 아닌지를 나타내는 클래스 레이블을 만들었습니다. NCBI에서 얻은 데이터는 34개의 Positive sample과 295개의 Negative sample로 구성됩니다.
모델은 Keras를 이용하였으며, CNN,Bi-LSTM, 출력층을 만들어 CNN-Bi-LSTM 모델을 만들었습니다. 배치 크기는 [관련 논문 12], [관련 논문 54]에서 언급한 대로 64개로 사용하였으며 Keras의 기본 Weight와 Bias를 사용했습니다. 모델은 권장되는 기본 학습률인 을 사용해 100 epochs동안 훈련되었습니다. LSTM의 셀 수를 32개부터 256개까지 탐색해 32개를 사용하였고, CNN의 필터 개수 역시 32개에서 256개까지의 탐색 후 기본값인 32개를 사용하였습니다. 또, binary cross-entropy와 Adam optimizer를 사용하였습니다. 이진 분류를 하기 위해 출력층의 active function으로 sigmoid함수를 사용하였습니다. 모델은 각 학습마다 다른 결과를 나타낼 수 있기에 각 실험을 100회 반복 후 100회 반복에 대한 평균값을 계산하였습니다.
Discussion
해당 논문에서 이루어진 성능 평가에선 CNN-Bi-LSTM이 Sens, Spec, Prec, Acc, AUC ROC의 지표에서 CNN과 Bi-LSTM모델과 유사한 성능을 달성했습니다. 또한, 이진 분류의 성능 측정에 사용되는 통계 측정인 MCC와 Cohen's Kappa에선 CNN과 Bi-LSTM보다 높은 점수를 산출하였습니다.
이 결과는 해당 모델이 confusion matrix의 네가지 항목에서 모두 우수한 결과를 얻었음을 나타냅니다.
또, AUC ROC에선 CNN 및 Bi-LSTM보다 높은 100%를 달성하여 CNN-Bi-LSTM이 이진분류에서 좋은 결과를 내는 것을 보입니다. 또, 세 모델 모두 P-value가 2.2e-16 < 0.05로 5%수준에서 유의미했으며, 이 결과는 분류 정확도가 NIR(No Information Rate)보다 상당히 높다는 것을 보여줍니다. 즉, 해당 모델이 불균형 데이터를 사용하는 경우에도 예측에 유용하다는 것을 보입니다.
CNN-Bi-LSTM 모델의 CNN층에선 DNA 시퀀스에서 가능한 많은 특징을 추출하며, Bi-LSTM층에서 과거와 미래의 상태를 학습하고, 시간적인 특징을 이용하여 예측에 사용합니다. Bi-LSTM은 긴 DNA Seq를 분석하는 데 중요한 데이터 사이의 시간 순서를 유지할 수 있습니다. 따라서 CNN을 Bi-LSTM의 입력에 이용하여 코로나 바이러스 중 SARS-CoV-2를 효율적으로 분류해 내는 하이브리드 모델을 개발하였습니다. CNN-Bi-LSTM 모델은 Max-Pooling 층과 연결되어 있는 3개의 Convolution층과 하나의 Bi-LSTM층, 분류를 위한 fully connected dense층과 100개의 뉴런을 가지는 fully connected neural network층으로 구성됩니다.
Convolution층은 32개의 kernel을 가지며, Bi-LSTM은 32개의 cell을 가집니다. 제안된 모델에서 Convolution층의 kernel 수와 Bi-LSTM층에서의 cell의 수가 증가하여도 유의미한 성능의 향상을 보이지 않았습니다. 관련 논문 19에 따르면 추가적인 convolution층과 max-pooling층의 사용 시 신경망이 더 깊어져 neural network의 학습을 저하시킵니다.
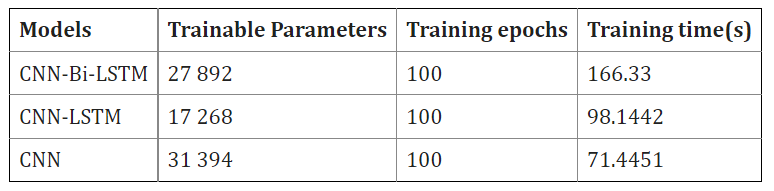
위의 표는 CNN의 전체 parameter 수가 CNN-Bi-LSTM이나 Bi-LSTM보다 더 많다는 것을 보여주며, 이에 따라 훈련 시간 역시 증가하는 것을 보여줍니다. CNN의 dense층은 이전 층의 모든 값에 연결되고, 이 연결을 매개변수화 하기 위해 더 큰 가중치 행렬이 필요합니다. 반대로, Bi-LSTM에선 feature map이 샘플 별로 반복적으로 처리되기 때문에 훨씬 더 적은 수의 parameter가 필요합니다. 즉, CNN-Bi-LSTM이 CNN에 비해 복잡한 모델임에도 불구하고, parameter가 더 적으며 이는 CNN-Bi-LSTM 모델 사용에 필요한 계산 리소스에 영향을 미칩니다.
제안된 모델에선 CNN에 Max-Pooling층을 사용해 down-sampling하여 모델이 학습해야 하는 parameter수를 줄임으로써 소요되는 훈련 시간을 줄였습니다. 또한, 해당 모델 학습 속도에 있어 learning rate나 Drop-out과 같은 매개변수가 학습 시간에 주는 영향이 미미함을 발견했습니다. 해당 발견은 관련 논문 67과 68에서 뒷받침 되고 있습니다.
또, 해당 연구에선 제안된 모델이 새로운 데이터에 적용되는 상황에서 역시 강력한 성능을 보인다는 것을 보였씁니다. 이는 비교적 적은 데이터로도 우수한 성능을 얻을 수 있다는 것을 보이며, 데이터의 수집에 많은 비용이 들 수 있는 상황에 해당 모델이 사용될 수 있음을 나타냅니다.
limitations
딥러닝 모델은 많은 수의 parameter들을 훈련해야 하기 때문에 많은 훈련시간을 필요로 합니다. 잘 훈련된 모델들은 많은 계산을 요구하며, 큰 메모리를 필요로 합니다. 때문에 모델의 배포는 계산 및 메모리 요구사항에 의해 방해받을 수 있습니다. 따라서 해당 논문에선 더 많은 계산자원을 필요로 하는 더 깊은 모델을 개발하지 않았습니다.
또한, 해당 논문에서 제안된 모델은 SARC-CoV-2와 다른 코로나 바이러스들을 분류하는 방법에 대한 설명을 제공 해 주지 않습니다. 즉, 해당 모델을 black box로 사용하였습니다.
그리고, 제안된 모델은 alignment-based method에 비해 더 많은 수의 훈련 데이터가 필요합니다.
하지만, 제안된 모델은 코로나 바이러스들 사이에서 SARS-CoV-2를 올바르게 분류하며, DNA Seq에 regulatory motif가 존재하는지의 여부를 확인할 수 있습니다.
해당 논문에선 향후 연구를 위해 Regulatory motif 구성 변경의 인과적 영향을 조사하고, 더 나아가 RNA-단백질 결합 예측을 탐구하기 위해 제안된 모델을 사용할 것을 권고하고 있습니다.
Reference
관련 논문 2
SARS-CoV-2와 다른 코로나 바이러스를 분류하는 연구
관련 논문 20
DNA binding motif를 예측하는 연구
관련 논문 24
DeapSEA 개발 연구
관련 논문 30
DanQ 개발 연구
추가적으로 공부 할 키워드
- alignment-based method
- Regulatory motif
- MCC
- Cohen's Kappa
- 5x2-fold cv paired t-test
- P-value
