SARS CoV-2 Deep Learning 논문 의 연구에서 해당 연구의 모델을 확장했기 때문에 해당 연구를 함께 리뷰합니다.
논문 리뷰에 앞서
논문 리뷰는 다회독을 하며 내용을 수정해 나갈 게시글입니다.
현재 1회독 중이며, Abstract, Introduction, Conclusion을 정리합니다.(22.10.04)
현재 2회독 중이며, 세부 내용을 정리합니다. (22.10.05)
리뷰 완료 (22.10.08)
1회차 리뷰
Abstract
오늘날 최소한의 전처리된 데이터에서 특성을 추출하는 CNN의 사용은 많아지고 있습니다.
해당 연구에서는 DNA Seq를 하나의 text data로 간주하여 CNN을 통해 븐류하는 접근법을 제시합니다.
Seq를 모델의 입력으로 제공하기 위해 One-hot vector를 사용합니다.
Introduction
DNA는 모든 생물의 발달, 기능, 생식에 대한 대부분의 유전적인 설명을 지니고 있기 때문에, DNA에 대한 연구는 생물의 이해에 매우 중요한 요소입니다.
시간이 지남에 따라 DNA (염기쌍)을 읽는 비용은 급격히 감소했으며, 염기 서열에 대한 데이터 양 또한 기하급수적으로 증가하고 있습니다.
이에 따라 컴퓨터를 이용하여 거대한 DNA데이터를 사용하는 것이 중요해 지기 시작했습니다.
해당 연구에서는 염기서열에서의 각 염기의 위치 정보를 보존하고 있는 seq를 one-hot vector로 나타내는 딥러닝 모델을 제안합니다.
해당 모델은 텍스트 분류 모델에서 영감을 받았다고 설명합니다.
Conclusion
CNN을 이용해 one-hot vector로 표현한 DNA Seq의 특성을 추출하는 것은 매우 좋은 성능을 보였습니다.
해당 모델을 이용하여 DNA Seq를 분석하는 다른 연구에 사용할 수 있을 것이라 언급합니다.
2회차 리뷰
Model
1. CNN
CNN이 사용 된 직관적인 연구는 얼굴 인식 어플리케이션의 모델이라 설명합니다.
해당 연구에서 사용된 모델은 다음 그림처럼 4개층의 Convolution Layer, 3개 층의 Fully connected Layer, Output Layer로 구성이 됩니다.
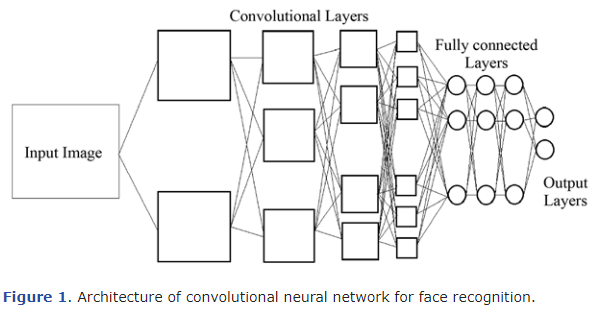
이 중 Convolution층에 대해 설명을 하는데, 4개 층 중 가장 첫번째 층은 주어진 pixel 입력에서 수직선, 수평선, 대각선 같은 매우 간단한 모양을 인식합니다.
이어지는 두번째 층에선 곡선이나 직선의 끝부분 등의 조금 더 복잡한 모양을 인식하고, 세번째 층에선 눈, 코, 입 등 사람 얼굴의 일부를 인식합니다.
마지막 층에선 사람 얼굴 전체를 인식합니다.
2. CNN for Text
위에서 설명했던 CNN은 주제 분류, 스팸 탐지, 감정 분류와 같은 Text data의 문제에서도 사용이 됩니다. 이전의 CNN에 입력된 2차원의 이미지 데이터와 달리 Text data는 연속적인 글자들로 이루어진 1차원 데이터 입니다.
다양한 연구에서 텍스트를 word vector라 불리는 텍스트 표현을 위한 벡터에 대응시키기 위해 Lookup table을 사용합니다.
( Lookup table이란 특정 연산에 대한 결과값을 미리 저장해 둔 배열 입니다. )
하지만 관련 논문 5의 연구에서는 lookup table을 사용하는 것은 unigram 정보를 사용하며, bigram이나 n-gram 정보를 활용하는 것이 샘플 분류에 더 효과적이라고 주장합니다.
( n-gram은 n개의 연속된 단어 Seq를 뜻합니다. 즉, unigram이란 하나의 단어 정보, bi-gram은 연속된 두개의 단어 정보, n-gram은 n개의 연속된 단어 정보를 뜻합니다. )
해당 연구에서는 lookup table을 사용하는 대신 단어를 표현하기 위해 one-hot vector를 사용하는 접근법을 제안합니다.
또, one-hot vector로 표현된 가까운 word vector들을 연결해 텍스트를 n-gram으로 나타냅니다.

위 그림에서는 텍스트를 one-hot vector를 이용해 2차원 행렬로 나타내는 과정을 설명합니다.
우선, 각 단어 I, cat, dog, have, a 에 대응되는 word vector 사전이 있다고 사정합니다.
이후, 주어지는 문장에 대해 n개의 연속된 단어들을 word vector로 표현하여 연결합니다.
위 그림의 경우 연속되는 단어의 개수를 2개로 지정하였습니다.
즉, I have a dog을 각각의 word vector 으로 표현 후 2개단위로 묶어서 위와 같이 2차원 행렬로 나타냅니다.
이렇게 생성된 2차원 행렬은 CNN에 입력되며, 이 모델을 seq-CNN 이라고 합니다.
이 모델은 감정 분류와 주제 분류 문제에서 성과를 거두었습니다.
3. CNN for Sequence
DNA-seq data는 text data와 달리 공백이 없는 글자 seq입니다.
즉, DNA-seq에서는 단어라는 개념이 존재하지 않기에 DNA-seq를 먼저 단어들의 seq로 변경해주어야 합니다.

위 그림은 DNA-seq를 단어들로 전환하는 과정을 설명합니다.
고정된 크기의 창을 정해진 보폭만큼 움직이면서 해당 창에 들어있는 글자들로 단어를 만듭니다.
그림에서는 3의 크기의 창을 1씩 이동시키면서 단어들을 만들어냅니다.
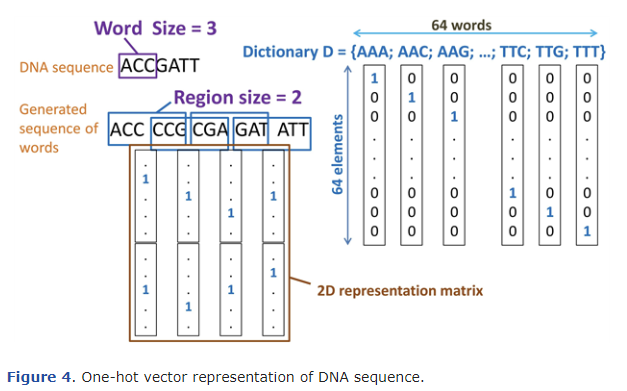
이렇게 생성된 1차원의 단어 Sequence를 seq-CNN과 같이 one-hot vector를 사용하여 2차원 행렬로 만들어 줍니다. 이 때 Nucleotide의 단어 개수는 총 64개 이므로, 사용되는 one-hot vector 역시 64의 크기를 가집니다. (각 Nucleotide의 종류는 4개이며, 3개의 Nucleotide가 하나의 단어를 구성하므로 개의 단어가 있습니다.)
이제 해당 행렬을 CNN에 입력해 분류를 수행합니다.
Experiments and Results
1. Datasets
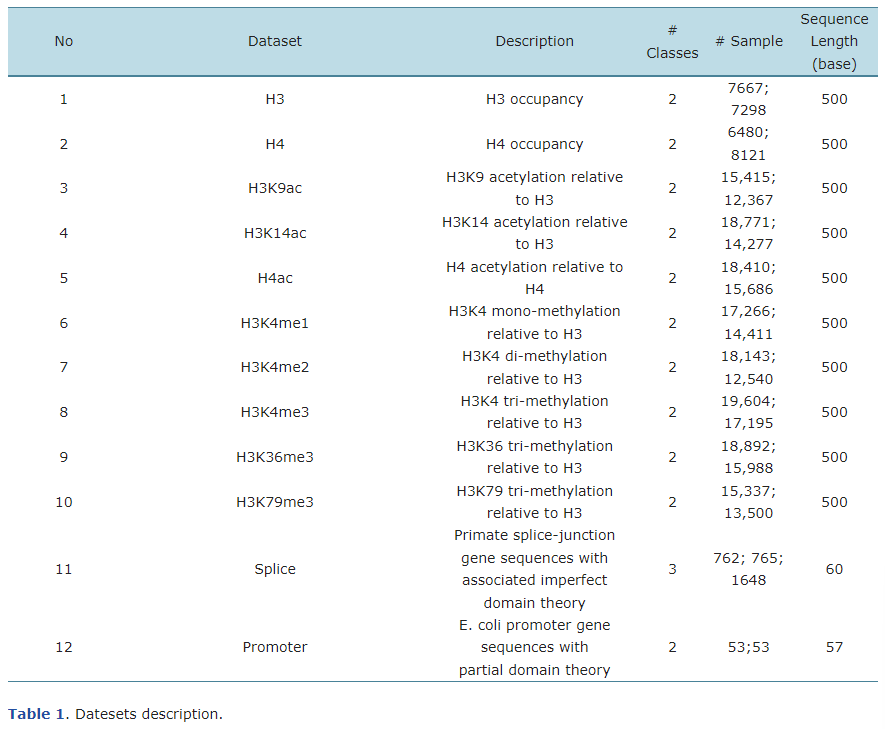
위의 표는 해당 연구에서 사용한 dataset들입니다.
1~10번의 데이터는 histone 단백질을 감싸고 있는 DNA Seq에 대한 데이터 이며, 관련 논문 6에서 도출 된 dataset 입니다. H3과 H4는 histone type을 의미하며, K와 그 뒤의 숫자는 변형된 아미노산을 의미합니다. (K14는 14번째 아미노산 K가 변경되었음을 뜻합니다.) ac, me는 변형의 종류(acetylation, methylation)을, me뒤의 숫자는 times of methylation을 뜻합니다.
해당 데이터는 500개의 염기쌍으로 구성되어 있으며, histone 단백질에 둘러쌓인 부분이 존재하는 Positive 클래스와 해당 부분이 존재하지 않는 Negative 클래스로 구분 됩니다.
11번 데이터는 Splice 데이터로 splice juction에 관한 데이터 입니다.
유존자에는 RNA 전사 과정에서 제거되는 intron이라 불리는 영역과 mRNA를 생성하는 데 사용되는 exons라는 영역이 존재합니다. 그리고 이 둘 사이의 접합부를 splice junction라고 부릅니다. splice juction는 Exon-Intron junction과 Intron-Exon junction의 두가지 종류가 있습니다.
Splice 데이터는 60개의 염기쌍으로 구성되어 있으며 Exon-Intron junction을 포함하는 EI 클래스, Intron-Exon junction를 포함하는 IE 클래스, 그리고 둘 다 포함하지 않는 N 클래스의 3가지 클래스를 가집니다.
마지막 데이터는 Promoter 데이터로 유전자 전사의 시작에 관여하는 유전자의 상류 영역인 promoter에 관한 데이터 입니다.
57쌍의 염기쌍으로 이루어진 promoter데이터는 promoter의 nucleotide를 포함하는 Positive 클래스와 해당 nucleotide를 포함하지 않는 Negative 클래스로 구성됩니다.
2. Model Configurations
해당 연구는 CONTEXT(GPU에서 text categorization을 하기 위해 CNN을 사용하는 C++ 소프트웨어 패키지)를 사용했습니다.
해당 모델은 2개의 convolutional layer를 포함하며 각 convolutional layer 뒤에는 sub-sampling layer가 뒤따릅니다. 해당 층들은 Seq의 행렬표현에서의 특징을 추출합니다.
추출 된 특징들은 100개의 뉴런을 가진 fully connected neural network layer를 사용해서 변환됩니다. 해당 층에서는 overfitting을 막기 위해 0.5의 dropout을 사용합니다.
마지막으로 soft max output layer를 통해 입력 seq의 레이블을 예측합니다.
3. Evaluation
모델의 성능 평가를 위해 10-fold cross validation을 3회 수행하여 평균 정확도를 계산했습니다. histone 단백질 데이터의 이전 최고 결과는 Higashihara의 2008년 연구 결과를 사용했습니다. Promoter 데이터 세트에선 이전 연구와 비교하기 위해 이전 연구와 같은 방법인 leave one out을 사용하였습니다. Splice 데이터에 대한 이전 최고 결과는 관련 논문 8를, Promoter 데이터의 이전 최고 결과는 Towell의 연구에 사용된 KBNN을 통해 사용한 결과를 사용했습니다.
4. Results
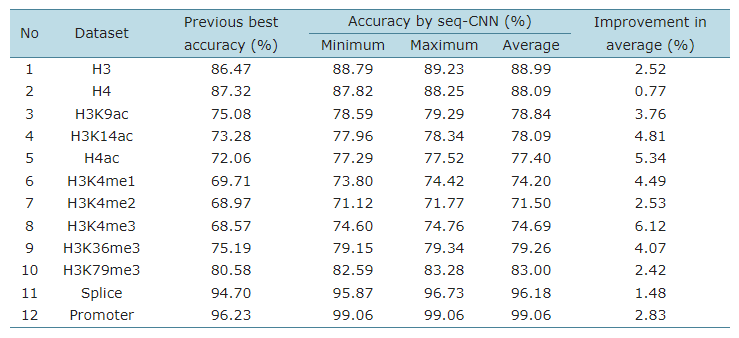
표 에서 볼 수 있는 것 처럼 모든 검증 데이터 세트에서 성능이 개선되었습니다.
이를 통해 CNN을 사용한 DNA-seq 분류가 유용하게 사용 될 수 있다는 것을 확인 할 수 있습니다.
Discussion
본 연구에선 단어 크기, 영역 크기 ,네트워크 구조 등 모델의 hyper-parameter를 경험에 기반해서 선택했으며 이에 대한 추가적인 연구가 필요하다고 언급합니다.
