3.1.5 수치형 변수를 정규분포 형태로 만들기
log 변환을 통해 전처리
- 학습 시 데이터의 분포가 skewed된 형태라면 어려움이 있음
- 한정된 케이스에 대해서만 학습하기 때문에... 이래서 balance가 중요
plt.figure(figsize = (10, 8))
sns.distplot(df.loc[df.Insulin>0, 'Insulin'])
plt.show()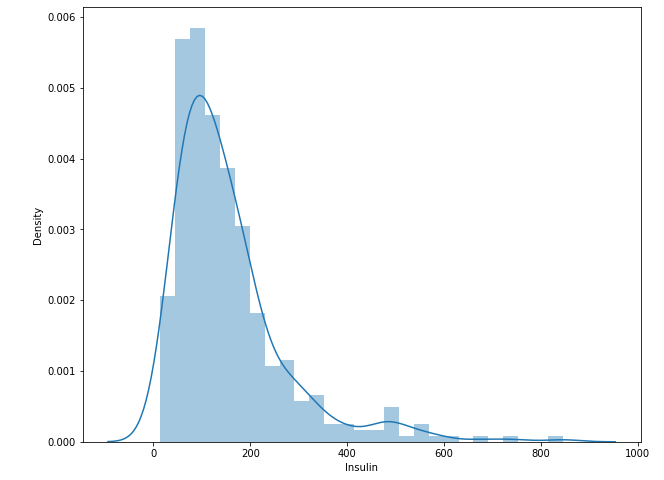
- 왼쪽으로 skewed 형태
- Log 변환을 해줘 shape의 변환!
plt.figure(figsize = (10, 8))
sns.distplot(np.log(df.loc[df.Insulin>0, 'Insulin']+1))
plt.show()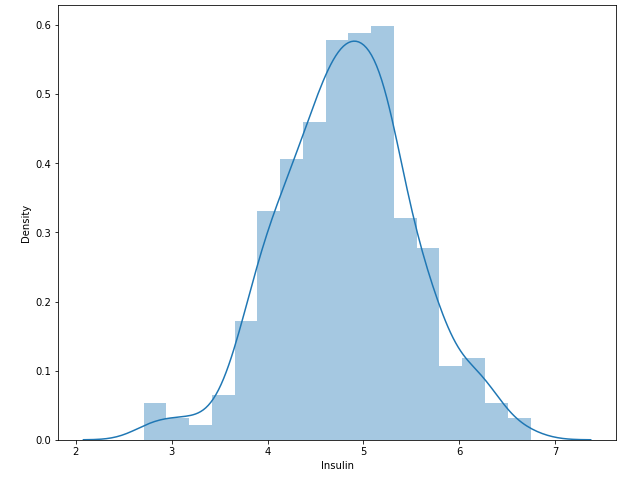
- log(0) = -Inf 이므로 +1을 해줌
- log 변환을 통해 꼬리가 긴 부분을 앞으로 당겨오고 전체적으로 scale 조정을 해 줌.
df['Insulin_log'] = np.log(df['Insulin_nan'] + 1)
fig, axes = plt.subplots(1, 2, figsize = (15, 5))
sns.distplot(df['Insulin_nan'], ax = axes[0])
sns.distplot(df['Insulin_log'], ax = axes[1])
plt.show()
- median으로 채워줬기 때문에 가운데가 뾰족한 형태
- 두 개의 봉이 생김
- 역시 log 변환 전에는 왼쪽으로 쏠린, 오른 쪽으로 꼬리가 긴 분포
- 앞으로 insulin_nan을 제거하고 학습하고 예측!
3.1.6 상관 분석을 통해 파생변수 만들기
피부 두께와 BMI의 상관성이 높았었음
plt.figure(figsize= (10, 8))
sns.lmplot(data = df, x = 'Insulin_nan', y = 'Glucose', hue = 'Outcome')
plt.show()
- 인슐린과 글루코스의 상관관계 파악
- 발병하지 않은 경우의 기울기가 더 가파름.
df['low_glu_insulin'] = (df['Glucose'] < 100) & (df['Insulin_nan'] <= 102.5)
df[['low_glu_insulin']].head()
- 인슐린 수치가 채워준 중앙값 이하, 글루코스 수치가 100 미만인 파생변수를 만들어 줌
pd.crosstab(df['Outcome'], df['low_glu_insulin'])
- 파생변수와 Outcome 간의 동등비교(==)를 한 결과를 tabel로 만들어 줌
- 파생변수가 True인 경우 발병한 경우는 5건. 상당히 낮음
features = df.columns.tolist()
features.remove('Pregnancies')
features.remove('Outcome')
features.remove('Age_low')
features.remove('Age_middle')
features.remove('Age_high')
features.remove('Insulin')
features.remove('Insulin_nan')
train = df[: int(df.shape[0] * 0.8)]
test = df[int(df.shape[0] * 0.8): ]
model.fit(train[features], train[label_name])
y_pred = model.predict(test[features])
diff_count = sum(abs(y_pred - test[label_name]))
print('Accuracy: {}'.format((len(y_test) - diff_count) / len(y_test)))
sns.barplot(y = features, x = model.feature_importances_)
plt.show()
- 정확도의 별 차이는 없고 새로 만들어낸 변수가 별 관계가 없음
3.1.7 이상치 다루기
fig, axes = plt.subplots(2, 1, figsize = (15, 6))
sns.boxplot(df.Insulin, ax = axes[0])
sns.boxplot(df['Insulin_nan'], ax = axes[1])
plt.show()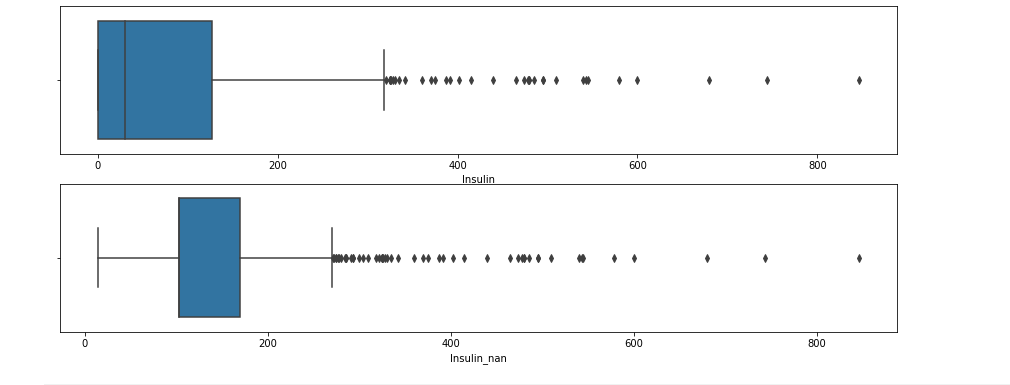
- 위가 nan 처리 전, 아래가 nan 처리 후.
- 분포가 더 좁은 형태이고 당연히 whisker도 더 작아질 수 밖에 없음
df['Insulin_nan'].describe()
- q1 == q2(median)인 이유는 0을 다 median으로 채워줬었기 때문
- max값이 q3에 비해 5배 가까이 크므로 이건 이상치에 가깝지 않을까?
### IQR
IQR3 = df['Insulin_nan'].quantile(.75)
IQR1 = df['Insulin_nan'].quantile(.25)
IQR = IQR3 - IQR1
IQR
- Pandas의
.quantile을 이용하면 원하는 분위수의 값을 얻을 수 있음. - 아니며 describe의 인덱스를 불러와서 값을 얻어도 됨.
- 아니면 정렬한 후에 원하는 분위수의 인덱스 값을 넣어서 인덱싱을 해도 됨.
OUT = IQR3 + 1.5*IQR
OUT
- box-plot에서 아래 수염보다 작은 이상치는 없었으므로 윗수염 기준으로 이상치 제거
- 270을 기준으로 보다 크다면 이상치로 처리
df[df['Insulin_nan'] > OUT].shape
- 전체 데이터가 700개 가량인데 51개를 제거해준다면 조금 양이 많음
- 따라서 600을 기준으로 이상치라 생각하고 결측치 처리
print(df[df['Insulin_nan'] > 600].shape)
train = df[df['Insulin_nan'] <= 600]
- 3건 뿐이므로 그냥 제외한다면
print(df[df['Insulin_nan'] > 600].shape)
train = train[train['Insulin_nan'] <= 600]
test = test[test['Insulin_nan'] <= 600]
features = df.columns.tolist()
features.remove('Pregnancies')
features.remove('Outcome')
features.remove('Age_low')
features.remove('Age_middle')
features.remove('Age_high')
features.remove('Insulin')
features.remove('Insulin_log')
model.fit(train[features], train[label_name])
y_pred = model.predict(test[features])
diff_count = sum(abs(y_pred - test[label_name]))
print('Accuracy:', (len(test[label_name]) - diff_count) / len(test[label_name]))
- 정확도가 87%. 그렇게 큰 차이는 딱히..
- 90%가 넘어가는 경우도 있었기 때문에 베스트가 아님
3.1.8 피처 스케일링
StandardSclaer
- 예를들어 x1은 단위가 km, x2는 m라고 하자. 100m를 표현하면 x2는 1, x1은 0.1이 된다.
- x2의 값은 x1보다 10배가 큰 경우가 되는데 이게 맞는 경우일까?
- 이처럼 피쳐별로 단위가 다르다면 같은 값이라 하더라도 그 영향이 달라질 수가 있다.
- 따라서, Sclaing을 통해 해당 값의 고유한 영향력만을 모델에 반영
from sklearn.preprocessing import StandardScaler
## z표준화 스케일러 생성
scaler = StandardScaler()
## 변환하고 싶은 df or array 넣어주고 fitting
scaler.fit(df[['Glucose', 'DiabetesPedigreeFunction']])
## 변환
scale = scaler.transform(df[['Glucose', 'DiabetesPedigreeFunction']])
scale
tmp = df[['Glucose', 'DiabetesPedigreeFunction']].copy()
tmp[['Glucose', 'DiabetesPedigreeFunction']] = scale
print(tmp.head())
## min-max scaling
tmp2 = df[['Glucose', 'DiabetesPedigreeFunction']].copy()
tmp2 = tmp2.apply(lambda x: (x - min(x)) / (max(x) - min(x)), axis = 0)
print(tmp2.head())
- 하나의 시리즈만 바꿀 수 있는게 아니라 df의 형태로도 바꿔줄 수 있음
- scaler의 transform 결과물은 array
h = df[['Glucose', 'DiabetesPedigreeFunction']].hist(figsize = (15, 3))
h2 = tmp.hist(figsize = (15, 3))
h3 = tmp2.hist(figsize = (15, 3))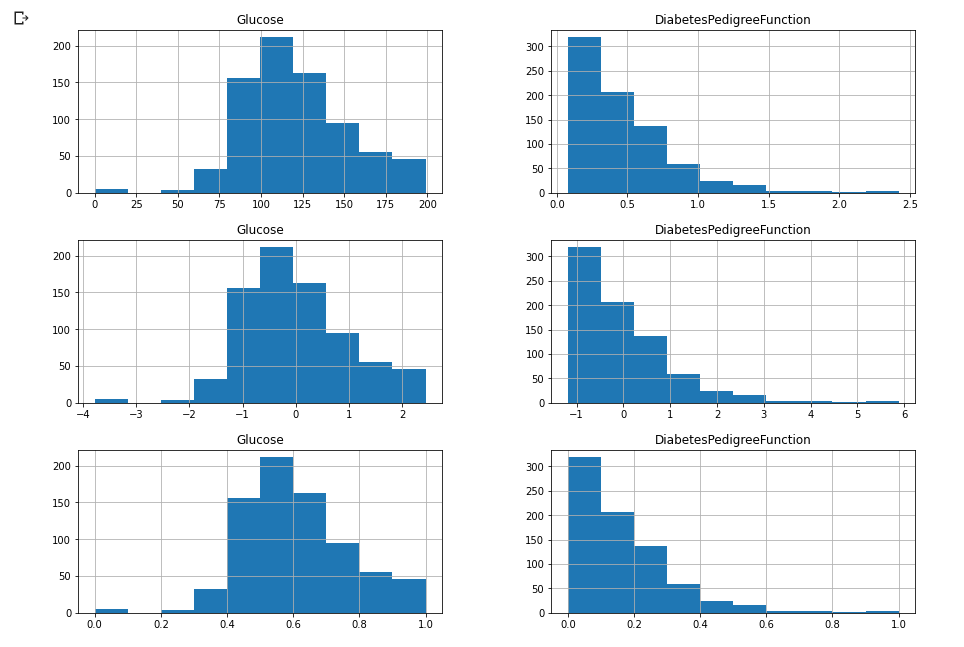
- 스케일링 하기 전과 후의 histogram
- mean을 빼주고 std로 나눠줌. 단위의 차이를 없애고 순수한 관측치의 영향만 남겨둠
- histogram의 형태는 둘이 똑같다고 생각됨
- scaling 자체가 값의 변화는 생겨도 분포의 변화는 없음.
이 프로젝트에선 스케일링이 큰 영향은 없음. 다른 프로젝트에선 정확도를 높일 수 있으므로 전처리는 EDA를 먼저 진행해 인사이트를 얻은 후 진행하는 것이 Good
3.1.9 전처리한 Feature를 저장하기
df.to_csv('/content/drive/MyDrive/edwith/프로젝트로 배우는 데이터 사이언스/data/diabetes_feature.csv', index = False)
pd.read_csv('/content/drive/MyDrive/edwith/프로젝트로 배우는 데이터 사이언스/data/diabetes_feature.csv').head()
- to_csv를 통해 원하는 경로에 원하는 파일명으로 내보낼 수 있고, index = False를 안준다면 컬럼의 0번째 위치에 인덱스가 같이 저장됨

Data science