1. 다중 서열 정렬(Multiple Sequence Alignment, MSA)란 ?
다중 서열 정렬(MSA) 는 셋 이상의 DNA 또는 단백질 서열을 가져와, 진화적으로 동일한 위치(상동 부위)를 찾아 나라히 정렬하는 분석 기법이다.
다중 서열 정렬의 주요 목적
- 진화적 관계 추정 : 여러 종에 걸쳐 보존된 서열 영역을 분석하여 계통 발생 관계(Phylogenetic relationship)을 추정하고 공통 조상을 찾는 데 사용된다.
- 기능적 부위 예측 : 여러 서열에서 공통으로 보존되는 단백질은 중요한 기능을 담당할 가능성이 높다.
- *단백질 3차 구조 예측 : 상동성 모델링(homology modeling)에서 알려진 구조의 단백질 서열과 구조를 모르는 단백질 서열을 정렬하여 구조를 예측하는 데 중요한 기초 자료로 사용된다.
- PCR 프라이머 설계 : 다양한 종의 특정 유전자를 증폭시키기 위해 공통적으로 보존된 서열 영역에 결합할 수 있는 축퇴 프라이머(Degenerate primer)를 설계할 수 있다.
2. 다중 서열 정렬 알고리즘
정확한 최적의 MSA를 찾는 것은 NP-Hard 문제 이기 때문에, 주로 경험적이고 근사적인 방법을 사용한다.
점진적 정렬(Progressive Alignment)
가장 널리 사용되는 방식으로, 모든 서열 쌍의 유사도를 계산하여 가이드 트리(Guide tree)를 만들고, 이 트리에 따라 가장 가까운 서열부터 차례대로 정렬을 추가해 나간다.
초기 정렬의 오류가 최종 결과에 계속 영향을 끼친다는 단점이 있다.
- 대표 도구 : ClustalW / Clustal Omega, T-Coffee
반복적 정렬 (iterative Alignment)
초기 정렬을 만든 후, 이를 반복적으로 수정하고 재정렬하여 점진적 방법의 단점을 보완하고 더 나은 정렬 결과를 찾아가는 방식이다.
- 대표 도구 : MUSCLE, MAFFT
3. Python으로 TP53 단백질 정렬하기
최신 트렌트 : MAFFT와 대화형 시각화
- MAFFT :
MUSCLE이나ClustalW보다 더 빠른 속도와 높은 정확도를 보여주는 MSA 프로그램으로, 현재 연구실에서 매우 선호된다. - 대화형 시각화 : 정적인 이미지 대신, 마우스로 직접 탐색하고 확대/축소하며 서열을 확인할 수 있는 대화형 시각화 라이브러리(
plotly)를 사용한다.
3-1. 분석 데이터 준비
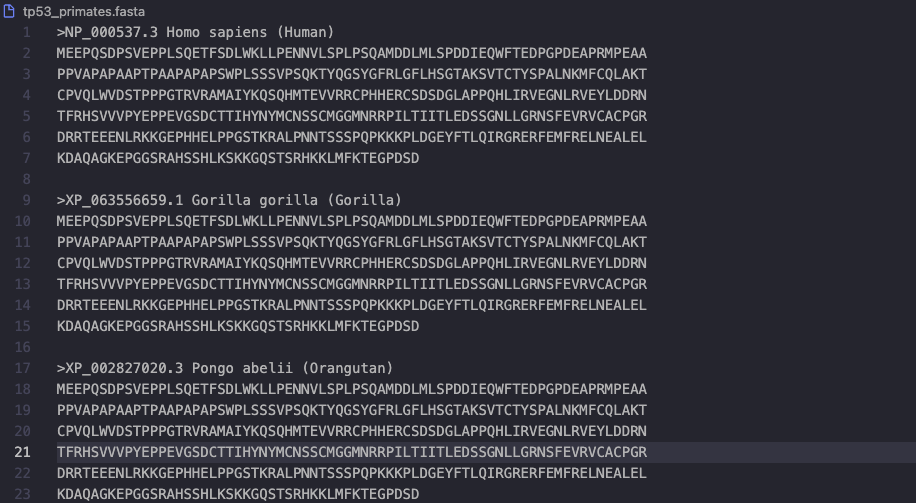
우선 사용 데이터는 인간, 고릴라, 오랑우탄의 TP53 단백질 서열을 사용할 것이며, 이를 NCBI에서 FASTA 형식으로 다운로드 하겠다.
from Bio import Entrez
Entrez.email = "bublman3375@knu.ac.kr"
# 가져올 단백질 서열의 NCBI id들
# 인간 , 고릴라, 오랑우탄
ids = ["NP_000537.3","XP_063556659.1","XP_002827020.3"]
output_filename = "tp53_primates.fasta"
print(f"NCBI에서 다음 ID의 서열을 다운로드합니다:\n{ids}")
try :
handle = Entrez.efetch(db="protein",
id=",".join(ids),
rettype="fasta",
retmode="text")
fasta_data = handle.read()
handle.close()
with open(output_filename,"w") as f :
f.write(fasta_data)
print(f"\n성공. '{output_filename}' 파일이 생성되었습니다.")
except Exception as e :
print(f"오류발생 : {e}")NCBI에서 다음 ID의 서열을 다운로드합니다:
['NP_000537.3', 'XP_063556659.1', 'XP_002827020.3']
성공. 'tp53_primates.fasta' 파일이 생성되었습니다.그리고 tp53_primates.fasta파일을 열어보면 데이터가 잘 받아와진 것을 볼 수 있다.
3-2. MAFFT와 Plotly를 사용하여 MSA 하기
우선적으로 plotly는 pip install plotly로 터미널에서 다운로드 받을수 있으며, MAFFT는 사이트에 접속하여 직접 다운로드 하여야한다.
https://mafft.cbrc.jp/alignment/software/
이제 필요한 라이브러리와 프로그램, 데이터가 준비되었으니 MSA를 해보도록하자.
from Bio.Align.Applications import MafftCommandline
from Bio import AlignIO
import plotly.express as px
import pandas as pd
import os
# --- 설정 부분 ---
input_file = "tp53_primates.fasta"
output_file = "tp53_aligned.fasta"
mafft_exe = "mafft"
# -----------------
try:
if not os.path.exists(input_file):
raise FileNotFoundError(f"입력 파일 '{input_file}'을 찾을 수 없습니다.")
print(f"MSA - MAFFT를 사용하여 정렬을 시작합니다...")
# --- 1. MAFFT 실행 ---
mafft_cline = MafftCommandline(mafft_exe, input=input_file)
stdout, stderr = mafft_cline()
with open(output_file, "w") as f:
f.write(stdout)
print(f"정렬 완료. 결과가 '{output_file}' 파일에 저장되었습니다.")
# --- 2. 정렬 결과 읽기 ---
alignment = AlignIO.read(output_file, "fasta")
aligned_sequences = [list(record.seq) for record in alignment]
sequence_ids = [record.id for record in alignment]
df = pd.DataFrame(aligned_sequences, index=sequence_ids)
# --- 3. [수정] 시각화를 위한 데이터 전처리 ---
# 각 아미노산 문자('M', 'E', 'P'...)를 고유한 숫자(0, 1, 2...)로 변환
print("\n시각화를 위해 아미노산 문자를 숫자로 변환합니다...")
# 모든 아미노산 종류를 찾아서 숫자와 매핑하는 딕셔너리 생성
amino_acids = sorted(list(set(df.values.flatten())))
aa_to_int = {aa: i for i, aa in enumerate(amino_acids)}
# 데이터프레임의 모든 문자를 숫자로 변환
df_numeric = df.replace(aa_to_int)
# --- 4. Plotly를 활용한 대화형 시각화 ---
print("Plotly를 활용하여 시각화를 시작합니다...")
fig = px.imshow(df_numeric, # !! 숫자 데이터프레임을 사용 !!
labels=dict(x="아미노산 위치", y="종(Species)", color="아미노산 ID"),
title="<b>TP53 단백질 다중 서열 정렬(MSA) 시각화</b>",
color_continuous_scale="Viridis")
# 마우스를 올렸을 때 원래 아미노산 문자가 보이도록 설정
fig.update_traces(
customdata=df.values, # 원본 문자 데이터를 customdata로 전달
hovertemplate="<b>위치</b>: %{x}<br><b>종</b>: %{y}<br><b>아미노산</b>: %{customdata}<extra></extra>"
)
fig.show()
except FileNotFoundError as e:
if "mafft" in str(e):
print("[에러] MAFFT 프로그램을 찾을 수 없습니다.")
else:
print(f"[파일 없음 에러] {e}")
except Exception as e:
print(f"알 수 없는 에러가 발생했습니다: {e}")4. 결과 분석

결과적으로 다음 그래프가 나오게 된다.
- X축 : 단백질을 구성하는 393개의 아미노산의 위치를 나타낸다.
- Y축 : 3종의 TP53 단백질 서열을 나타낸다.
그래프의 모든 열이 위아래로 완전히 동일한 색상 패턴을 보이는것은 "인간, 고릴라, 오랑우탄의 TP53 단백질은 아미노산 서열이 100% 동일하다" 라고 결론 지을 수 있다.
수백만 년에 걸쳐 인간, 고릴라, 오랑우탄이 공동 조상으로부터 현재까지의 진화 과정속에서 이 단백질의 서열에는 어떤 변화도 허용되지 않았음을 의미한다.
또, 해당 유전자에 돌연변이가 발생했다면, 그 개체는 암 발생 억제 능력이 떨어져 생존에 불리하여 도태되었다는 음성 선택(Negative Selection) 가설을 강력하게 뒷받침 한다.
