단일 아미노산 서열만을 기반으로 3차원 단백질 구조를 예측하는 새로운 모델 RGN2
🧠 배경 및 필요성
기존 구조 예측 모델들(예: AlphaFold2)은 고정밀 예측을 위해 MSA (Multiple Sequence Alignment) 기반 공진화 정보에 의존함. 하지만 이는 다음과 같은 한계를 가짐:
서열 상 동족(homolog)이 없는 고아 단백질(orphan proteins) 은 예측이 어려움.
de novo 설계된 단백질은 공진화 정보가 존재하지 않음.
MSA 생성은 계산 비용이 높고 느림.
👉 따라서 단일 서열만으로도 빠르고 정확하게 구조를 예측할 수 있는 새로운 방법이 필요함.
🧬 제안된 모델: RGN2 (Recurrent Geometric Network 2)
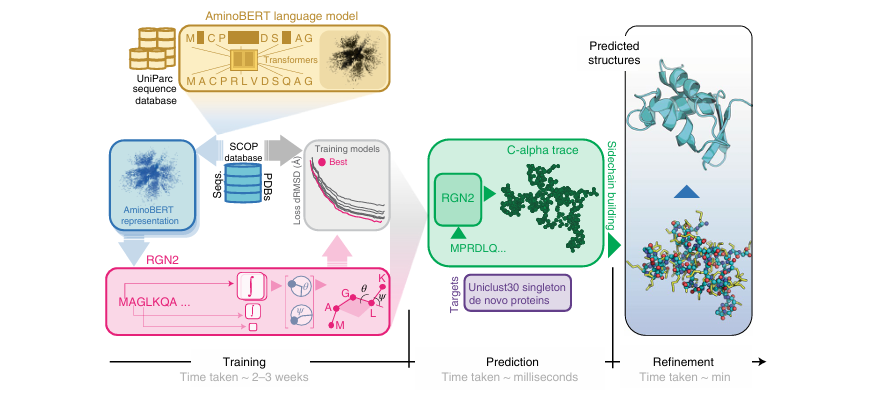
transformer 기반 protein language model (AminoBeRT, 노란색)
Frenet-Serret 프레임을 사용하여 단백질의 백본 구조를 생성하는 RGN(녹색)
side chain 과 수소 결합 네트워크의 초기 구축 후, AF2Rank(파란색)를 사용하여 구조를 정밀화
✅ 주요 구성
AminoBERT:
단백질 서열을 입력받아 구조 관련 정보를 추출하는 Transformer 기반 단백질 언어 모델
(MSA 사용 X)
약 2.5억 개 단백질 서열로 사전학습됨 (UniParc 데이터)
AminoBERT는 아미노산 서열에 내재된 구조 정보(접힘 규칙 등) 를 학습함.
이는 공진화 없이도 서열 자체로부터 구조를 유추한다는 뜻.
RGN1은 PSSM을 사용했기 때문에 MSA (동족 서열 정렬)가 필요했음.
RGN2는 단일 아미노산 서열만을 입력으로 사용하여 고아 단백질 / 설계 단백질에도 적용 가능.
AminoBERT는 BERT와 달리 아래 두 가지 추가 task 를 사용
연속된 Clumped Masking (2~8개) →
한 번에 2~8개의 연속된 아미노산을 마스킹 →
문맥을 더 넓게 학습하도록 유도.
chunk permutation (단편 재배열) →
서열을 여러 조각으로 나눈 뒤 순서를 섞음 →
전체 구조적 일관성 파악을 학습.
기하학 모듈 (Geometry Module):
Frenet–Serret 공식을 활용하여 Cα trace를 회전·이동 불변 형태로 표현
기존 RGN1은 torsion angle을 사용했지만, 이 방식은 때때로 물리적으로 불가능한 값을 생성함.
RGN2는 Frenet–Serret frame이라는 회전 및 이동 불변 구조 표현을 사용함 → 더 물리적으로 타당하고 계산 효율성 높음.
이는 용액 상태의 폴리펩타이드의 핵심 특성
단백질 주사슬의 Cα trace를 1차원 곡선으로 보고,
각 위치에 대해 Frenet–Serret frame 을 정의.
기존 RGN1이 사용하던 torsion angle보다 물리적으로 더 타당하고 계산 효율성 향상
🧪 성능 평가
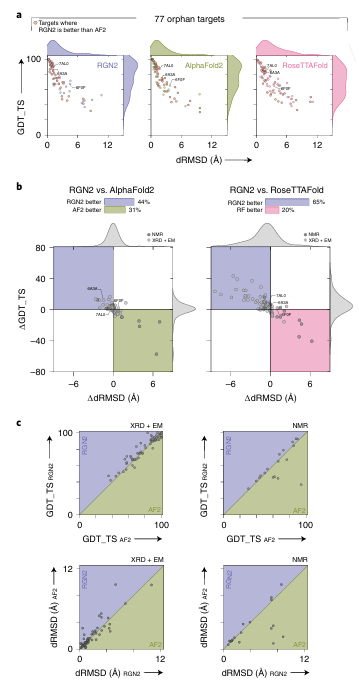
- Orphan 단백질에서의 구조 예측 성능 비교
a. MSA가 존재하지 않는 77개의 orphan 단백질
X축: dRMSD (Å) — 낮을수록 더 정확한 구조 예측
Y축: GDT_TS — 높을수록 더 정확한 전체 구조 매칭
b. RGN2 vs AF2/RF: 상대 성능 비교
X축: ΔdRMSD (RGN2 – AF2) → 왼쪽으로 갈수록 RGN2 우세
Y축: ΔGDT_TS (RGN2 – AF2) → 위로 갈수록 RGN2 우세
왼쪽 위 (보라): RGN2가 두 지표 모두 우세한 경우
오른쪽 아래 (초록/핑크): AF2 or RF가 우세
c. 예측 정확도 vs 실험 방법
GDT_TS / dRMSD 성능을 NMR/X-ray 기준으로 나누어 비교
대각선 위: RGN2 우세 / 아래: AF2 우세
NMR 기반 구조 (작은 단백질, 유연성 있음)에서 RGN2가 AF2보다 더 잘 예측하는 경향
X-ray 기반 구조에서도 많은 케이스에서 RGN2가 더 좋거나 비슷함
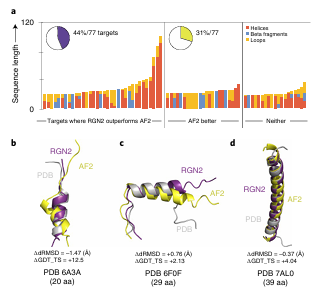
a. 단백질 길이 & 구조 요소별 분포
막대그래프: 각 orphan 단백질의 길이 + 구조 요소 비율
세 가지 그룹으로 나눔:
RGN2 우세 (보라)
AF2 우세 (초록)
우열 없음 (회색)
RGN2가 잘 예측한 단백질은 알파-헬릭스 구조가 많음
AF2는 베타 시트가 많은 단백질에서 상대적으로 강함
특히 길이가 긴 알파-헬릭스 단백질에서 RGN2가 더 뛰어남
이유: RGN2의 Frenet–Serret 기하 표현은 곡률·비틀림만 필요함 → 헬릭스 구조에 내재적으로 잘 적합
b–d. 구조 시각화 예시
알파-헬릭스 구조를 가진 orphan 단백질
RGN2는 헬릭스 구조 위주 단백질에서 뛰어난 성능
AF2는 베타 시트 구조에서 우세
- De novo 단백질 구조 예측
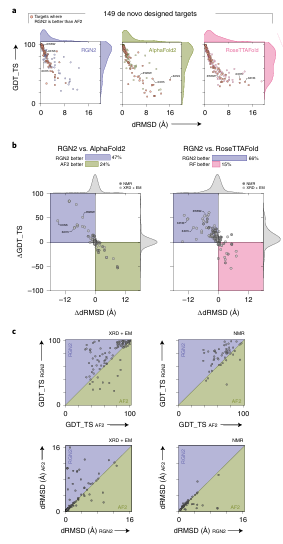
▶ 지표: dRMSD + GDT_TS
dRMSD: 구조 예측 정확도 (낮을수록 좋음)
GDT_TS: 전체 구조 매칭 정도 (높을수록 좋음)
AF2와 RF 모두 RGN2보다 느리고 예측력 떨어짐
특히 RF는 de novo 설계용으로 많이 사용되는데도 성능 밀림
실험 구조(X-ray/EM 기반 66%)로만 평가해도 유사한 결과
ΔGDT_TS: +15.5~16.9
ΔdRMSD: –2.38~2.65 Å
✅ (a) GDT_TS vs dRMSD 절대 성능
RGN2 (보라), AF2 (초록), RF (핑크)
RGN2가 대체로 더 높은 GDT_TS + 낮은 dRMSD → 더 좋은 예측 품질
✅ (b) RGN2 vs AF2, RF 비교 (ΔdRMSD vs ΔGDT_TS)
왼쪽 위 (보라 사분면): RGN2가 두 지표 모두 우세
149개 중 47%에서 AF2보다, 66%에서 RF보다 더 좋음
✅ (c) 실험 구조 유형별 비교 (XRD vs NMR)
X-ray / EM 구조 기반의 경우에도 RGN2 우세가 분명함
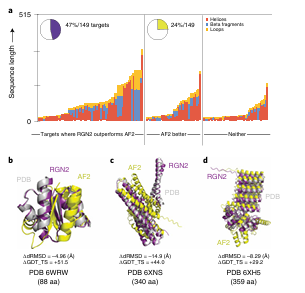
RGN2는 특히 알파-헬릭스 기반 설계 단백질에서 뛰어난 예측 성능
반면, 베타 시트가 많은 구조는 여전히 어려움
✅ (a) 단백질 구조 요소 분포
RGN2 우세 단백질: 헬릭스 구성 비율 높음
AF2 우세 단백질: β-fragment, loops가 상대적으로 많음
예시 구조 분석 (Fig. 5b–d)
2️⃣ RGN2 Prediction Speed (예측 속도)
MSA 없이 단일 서열로 작동 → MSA 계산 시간 없음
RGN2는 최대 10⁶배 빠름
⚠️ 한계 및 미래 방향
-
베타 시트 예측 정확도는 MSA 기반 모델보다 낮음
-
국소 정보에 집중
현재는 Cα 간의 국소적인 관계 (curvature, torsion)에만 의존 → 장거리 상호작용 반영 부족