💡 INTRODUCTION
✅ 무엇을 하는 서버인지
DUET은 단일 아미노산 치환이 단백질의 안정성(ΔΔG) 에 미치는 영향을 예측하는 웹 서버
서로 관점이 다른 두 방법
mCSM(구조 기반 그래프 서명)과 SDM(환경-특이적 치환표에 기반한 통계 퍼텐셜)
의 출력을 서포트 벡터 머신(SVM) 으로 통합해 합의(prediction consensus) 값을 내는 게 핵심 아이디어
이렇게 하면 개별 방법 하나만 쓸 때보다 전반적인 정확도가 좋아진다고 주장
✅ 왜 두 방법을 합치나?
과거 15년간 여러 in silico 예측법이 나왔지만 모든 상황에서 항상 정확한 단일법은 없다는 문제 제기
mCSM과 SDM은 서로 보완적(서로 다른 단백질 구조 특성/진화 정보 활용)이라서 통합 예측이 개별 예측보다 더 낫다는 논리
✅ 학습 데이터와 성능
ProTherm의 실험 열역학 데이터로 훈련했고,
블라인드 테스트로 검증했을 때 피어슨 상관계수 r ≈ 0.74(훈련), 0.71(테스트) 수준의 성능을 보고
(상위 10% 이상 편차치 제거 시 r이 더 올라감.)
또한 7개 방법을 묶은 다른 메타-예측기(iStable) 와 비교해 DUET이 더 일관되게 우수
✅ 의의(왜 유용한가)
대규모 유전체 프로젝트가 양산하는 nsSNP 들의 구조/기능 영향을 빠르게 가늠하고,
질병 연관성 분석이나 단백질 공학(설계·최적화) 에 활용할 수 있는 스케일러블한 도구
💡 MATERIALS AND METHODS
✅ SDM (Site-Directed Mutator): 환경-특이적 치환표 기반 통계 퍼텐셜
무엇을 학습했나:
동족 단백질군에서 관찰되는 환경-특이적 치환 경향(environment-specific substitution tables)을 통계 퍼텐셜로 사용해, 잔기의 즉시 주변 환경이 부여하는 진화적 제약을 반영한다는 가정
ΔΔG 계산 논리:
WT와 변이체를 각각 접힌 상태/풀린 상태에서의 아미노산 선호도(퍼텐셜)를 비교하여 접힘 자유에너지 변화(ΔΔG) 를 추정.
즉, “해당 환경에서 이 치환이 얼마나 자연스럽나/부자연스러운가”를 에너지 차로 환산한다는 관점
실무 디테일:
SDM은 변이체 구조가 필요해서 서버가 ANDANTE로 변이 구조 PDB를 만들어 쓰도록 되어 있음
또한 RSA(상대 용매접근도) 를 써서 회귀 트리(M5P) 로 SDM 기본 예측을 사전 보정(r 0.56→0.62)한 뒤, 통합 단계로 넘김
✅ mCSM (mutation Cutoff Scanning Matrix): 구조 기반 그래프(거리 패턴) 서명 + ML 회귀
표현 방식:
WT 구조에서 변이 잔기 주변을 그래프로 보고, 거리 패턴(Cutoff Scanning Matrix, CSM)과 파마코포어(원자 특성) 분포로 3D 화학 환경 서명을 만듦
이 서명을 지도학습 회귀에 넣어 변이가 안정성에 미치는 효과(ΔΔG)를 직접 예측
파마코포어 벡터:
DUET은 mCSM 쪽에서 WT↔변이 간 8가지 원자 특성(hydrophobic, ±전하, H-bond donor/acceptor, aromatic, sulphur, neutral)의 빈도 변화를 요약한 벡터도 같이 사용
✅ DUET – 통합 계산 접근(Integrated Computational Approach)
SVM 통합(스태킹) : “비선형 합의값” 만들기
무엇을 통합하나: 단일 점변이가 주어지면, mCSM + SDM의 예측을 비선형으로 결합해서 합의값(consensus) 을 내는 SVM 회귀(RBF 커널) 모델이 핵심
보완 피처 추가:
2차 구조(SDM 측정치),
WT↔변이 간 변화(원자 특성 8종: hydrophobic/positive/negative/H-acceptor/H-donor/aromatic/sulphur/neutral)를 요약한 파마코포어 벡터(mCSM에서 계산)도 함께 사용
입력 피처(스택의 상층):
mCSM의 ΔΔG 예측값,
(RSA로 보정된) SDM의 ΔΔG 예측값,
2차 구조 정보(SDM 쪽),
파마코포어 벡터(mCSM 쪽).
이 피처들을 SVM 회귀(RBF 커널) 에 넣어 최종 ΔΔG를 냄
왜 SVM인가:
두 베이스 예측기(SDM·mCSM)는 서로 다른 정보원(진화적 치환 경향 vs 3D 환경-기반 ML)을 봄
SVM(RBF)은 두 값의 신뢰도/오차 상관을 비선형으로 학습해 가중치를 조절하고,
보조 피처(2차 구조, 파마코포어)로 문맥 의존적 교정을 하며,
특정 영역(예: 노출·매몰, 구조 요소)에 따라 각 방법의 강점을 선택적으로 반영할 수 있음
그 결과 단독법보다 상관↑, 표준오차↓가 일관되게 나타남
(예: blind-set1에서 DUET r=0.71 vs SDM 0.56 / mCSM 0.67).
SDM 보정(필터링) 단계: 잔기 상대 용매접근도(RSA) 를 이용해 회귀 모델 트리(M5P) 로 표준 SDM 예측을 먼저 최적화한 뒤 mCSM과 결합
이 보정만으로도 블라인드 테스트에서 상관 r=0.56 → 0.62로 향상
최종 결합: (1) mCSM 예측, (2) 보정된 SDM 예측, (3) SDM의 2차 구조, (4) mCSM의 파마코포어 벡터를 SVM 에 넣어 최종 ΔΔG 예측을 출력
학습·검증은 각 변이에 대한 실험 열역학 데이터로 수행
SDM은 “이 환경에서 이런 치환은 진화적으로 자연스러웠나?”를 통계로 보는 룰-기반 척도
mCSM은 “주변 원자들과의 기하·화학적 패턴이 어떻게 바뀌는가?”를 데이터-기반으로 학습
SVM 통합은 두 시각을 하나의 비선형 함수로 결합해, 상황별로 더 믿을 수 있는 쪽에 가중치를 실어 최종 ΔΔG를 뽑는 스태킹 메타-모델
💡 WEBSERVER
✅ 입력(Input)
PDB 파일 또는 단백질의 4-letter PDB 코드 + 변이 정보(잔기 번호, WT/변이 아미노산 1-letter, 체인 ID)
스캔 기능: 특정 위치를 19개 모든 아미노산으로 한 번에 치환하는 systematic mutation 옵션 제공
구조 유형: NMR 구조도 지원하지만 첫 번째 모델만 사용
업로드 권장: 가능하면 단일 체인 PDB를 권장(결합 시 접히는 IDP 등은 예외)
✅ 출력(Output)
mCSM, SDM의 개별 예측값과 DUET의 통합(합의) 예측값, 그리고 GLMol 기반 인터랙티브 뷰어
ΔΔG(접힘 자유에너지 변화) 로 표시
도식 설명은 ‘양수 = 불안정화, 음수 = 안정화’
보조 정보: RSA(Richards 방법), 사이드체인 수소결합 만족도, 이차구조(SSTRUC) 도 함께 계산·표시
💡 VALIDATION
✅ 검증(Validation) — 데이터셋
학습 데이터: ProTherm에서 모은 변이 열역학 값 기반
학습셋은 S2848(PoPMuSiC가 사용한 컬렉션)에서 무작위 2,297개 변이를 추출해 구성
블라인드 테스트셋: 과적합을 줄이기 위해 2개의 blind set을 설계.
그중 첫 번째는 position 비중복 351개 변이로, 같은 위치의 변이는 학습/테스트 중 오직 한쪽에만 포함되도록 함
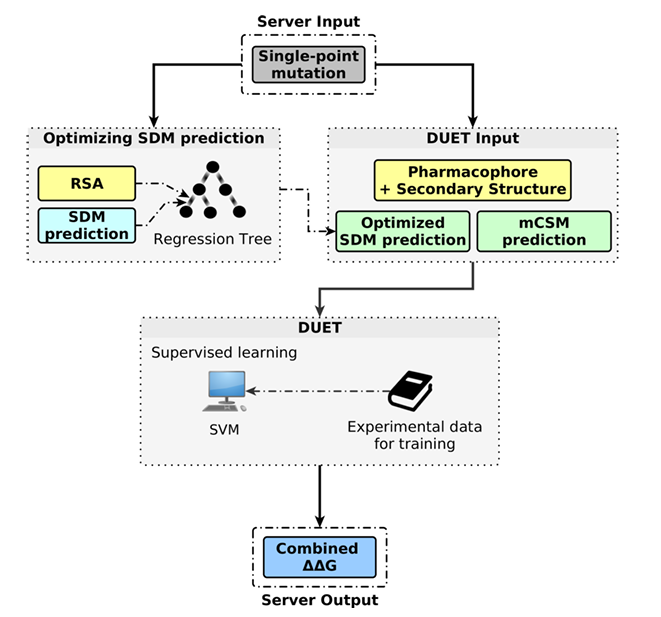
✅ 워크플로
입력: 단일 점 변이(single-point mutation)
SDM 보정 단계: 변이 잔기의 상대 용매접근도(RSA) 를 이용해 회귀 트리(M5P) 로 SDM 예측을 먼저 최적화(optimized SDM prediction)
DUET 입력 특성:
- mCSM 예측값,
- 최적화된 SDM 예측값,
- 2차 구조(SDM),
- 파마코포어 벡터(mCSM, WT↔변이의 8종 원자 특성 변화)
결합 모델: 위 특성들을 SVM 회귀(RBF 커널) 에 넣어 통합 ΔΔG를 출력(지도학습은 실험 열역학 데이터로 학습)
💡 RESULTS
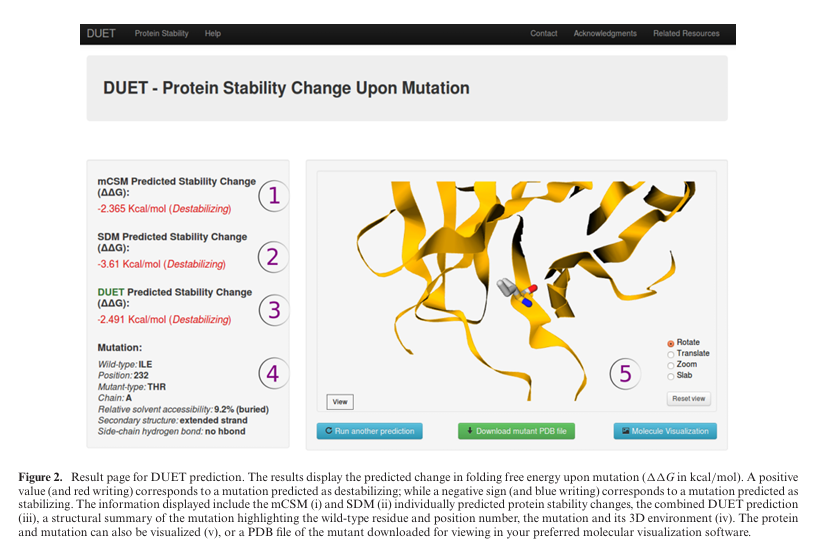
Figure 2 (결과 페이지 예시):
ΔΔG(접힘 자유에너지 변화, kcal/mol)을 표기.
양수(빨강)=불안정화, 음수(파랑)=안정화.
mCSM, SDM 단독 값과 DUET 통합값, 변이의 3D 환경 시각화, 변이체 PDB 다운로드 제공
✅ 성능 요약(본문에서 바로 이어지는 결과)
학습 성능:
DUET 회귀모델은 학습 동안 r = 0.74, σ = 0.98 kcal/mol.
단독 mCSM(r = 0.69, σ = 1.06)보다 유의하게 우수.
10% 이상치 제거 시 r = 0.82, σ = 0.72.
블라인드 테스트 1 (351 변이):
r = 0.71, σ = 1.13.
개별법 성능(SDM r = 0.56, mCSM r = 0.67)보다 높음.
상위 90% 구간에서는 r = 0.79, σ = 0.84.
✅ iStable과의 비교(p53 데이터셋)
데이터셋: 문헌에서 열역학 효과가 보고된 p53 DNA-binding domain의 42개 변이(학습셋과 중복 없음)
비교 결과(표 1):

p53 비교(42 변이)
표의 수치(요약):
(각 열의 앞/뒤 값은 “전체/이상치 10% 제거” 순).
mCSM: r 0.68 / 0.72, SE 1.40 / 1.20
SDM: r 0.52 / 0.64, SE 1.61 / 1.32
iStable: r 0.49 / 0.64, SE 1.59 / 1.37
DUET: r 0.68 / 0.76, SE 1.39 / 1.13
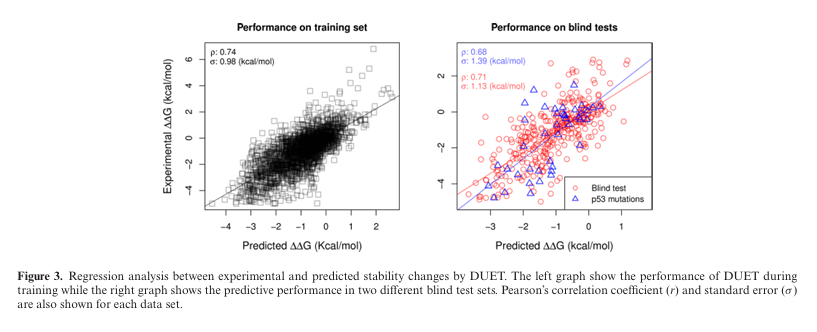
Figure 3 (회귀 산포도):
왼쪽은 학습셋, 오른쪽은 두 개의 블라인드 셋 성능(r, σ)을 각각 표시.
한 줄 결론
두 방법(SDM, mCSM)을 SVM으로 합의한 DUET은 학습·블라인드 테스트·p53 비교 모두에서 단독/타 메타예측기보다 일관되게 더 높은 상관과 더 낮은 표준오차를 보임