접힌 상태뿐 아니라 ‘풀린(unfolded) 상태’를 명시적으로 모델링해서 단백질의 절대 접힘 안정성(ΔG)을 예측하는 딥러닝 모델 IFUM을 제안
ΔG와 접힘/풀림 상태의 평형 앙상블(distogram)을 공동 학습해서 정확도를 끌어올린 게 포인트
💡 Abstract
기존 방법(구조예측 AI 기반 지표 포함)은 정량적 ΔG 재현에 한계가 있었음
IFUM은 ΔG와 접힘/풀림 상태의 잔기쌍 거리 확률분포(= distogram)를 함께 예측하여 단독 ΔG 학습보다 예측 성능을 크게 향상
데이터셋을 확장(메가스케일 소형 단백질, 무질서 단백질 포함 학습, 최대 869 aa 자연 단백질로 검증)하여 단백질 크기·종류 전반에 강건
또한 삽입/결실까지 포함한 일반화된 변이 효과(ΔΔG)도 잘 예측
실제 설계 2건:
(1) 다중 치환/삽입이 많은 블라인드 엔지니어링에서 실험 Tm와 양호한 상관
(2) de novo 설계 선택에서 널리 쓰이는 AF 기반 지표보다 현저히 향상
💡 Introduction
ΔG 실험 측정은 어렵고 비싸며 조건 이질성 때문에 비교도 어려움
→ 계산 예측의 필요
하지만 pLDDT/pTM 같은 구조신뢰도 지표나 PLM 기반 점수는 ΔG 자체를 정량 예측하도록 학습되지 않아 한계
(안정성으로 직접 학습된 게 아니라 필터 용도로 쓰일 뿐, 정량 ΔG를 내기엔 한계)
기존 ΔΔG 예측도 주로 점 돌연변이(single point)에 국한
(절대 안정성(ΔG)이나 삽입/결실엔 약함)
저자들은 문제의 근원을 “풀린 상태에서의 서열→구조 관계가 모호”하기 때문이라고 봄
ΔG = G_folded − G_unfolded 이므로 풀린 상태를 함께 고려하는 게 자연스럽다는 논지
과거엔 풀린 상태를 선형 ‘reference state energy’로 간단히 대체하거나 아예 null state로 취급
따라서 풀린 상태를 DL 아키텍처 안에 ‘명시적으로’ 추상화하는 것이 핵심 과제라고 제시
✅ IFUM의 개념적 가정 2가지
(1) 단백질은 two-state folding(접힘/풀림)으로 본다
(2) 풀린 상태 앙상블은 Flory random coil로 근사한 평균 거리 맵(ensemble-averaged distogram)으로 단순화할 수 있다
(고분자 물리에서 임의보행 평균 거동을 쓰는 고전적 모델)
풀린 상태의 평균 거리(Flory)와 접힌 상태의 실제 거리(ESMFold)를 ΔG로 정해지는 가중치로 섞어, 실제 조건에서의 잔기쌍 거리분포(평형 앙상블)를 만들고 이를 예측하도록 학습
→ 이 원리에 따라 IFUM은 접힘/풀림 상태를 잔기쌍 거리 맵으로 표현하고, ΔG = −RT ln(unfolded/folded) 정의에 부합하도록 열역학적 선호도와 함께 ΔG를 추정
삽입/결실을 포함한 보다 일반적인 ΔΔG도 다룰 수 있음을 보임
즉, ΔG 로 얻어낼 수 있는 가중치 값으로, folded 상태의 실제 거리는 알고 있으니, 이 두 개의 값을 통해, unfolded 의 거리를 계산하자 라는 뜻
🖐 “접힘(F) ↔ 풀림(U) 두 상태가 평형을 이룬다”는 뜻
실제 단백질은 수많은 미세한 구조(마이크로상태) 사이를 계속 흔들리지만, 큰 덩어리로 보면 ‘접힌 상태(F)’ 집합과 ‘풀린 상태(U)’ 집합 두 거대상태가 같은 조건(온도·완충액 등)에서 일정한 비율로 공존한다는 두-상태 가정(two-state assumption)
이때 두 상태의 양(점유율) 비는 자유에너지 차이 ΔG로 결정
RT를 곱해 자유에너지와 점유비(농도비)를 연결해 주는 관계식
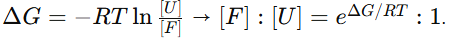
여기서 [F]와 [U]는 접힘/풀림의 평형 농도(점유율)
R: 기체상수(=몰 볼츠만 상수) -> 8.314 J·mol⁻¹·K⁻¹ = 1.987 cal·mol⁻¹·K⁻¹ = 0.001987 kcal·mol⁻¹·K⁻¹
T: 절대온도(Kelvin)-> 실온 25 °C는 298 K.
RT(25 °C) ≈ 8.314×298 ≈ 2477 J/mol = 2.48 kJ/mol
0.001987×298 ≈ 0.592 kcal/mol
접힘 분율 𝛼
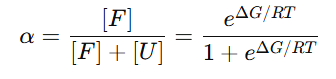
즉, ΔG가 클수록(접힘이 유리할수록) α↑, ΔG가 작거나 음수면(풀림 유리) α↓
직관적 예(25 °C 가정)
RT≈0.592 kcal/mol.
ΔG = 0 → α=0.50 (반반)
ΔG = 3 → α≈0.994 (거의 전부 접힘)
ΔG = −1 → α≈0.156 (대부분 풀림)그래서 한 조건에서 단백질은 한 가지 모양만 있는 게 아니라, 접힘/풀림 두 무리의 혼합비가 ΔG로 정해진다고 보는것
(이 혼합비를 논문은 나중에 거리분포 가중치로 쓴다.)
🖐 “무수한 풀림 구조들을 Flory random coil로 평균 거리(distogram)로 단순화”
왜 이런 단순화가 필요?
풀린 상태는 모양이 너무 다양해서 하나하나 모델링하기 어려움
그래서 고분자물리의 Flory random coil을 써서, 사슬 길이만으로 기대되는 평균적 거리 스케일을 주는 통계적 모델로 묶어 표현
어떻게 거리(레이블)를 만들었나?
풀림(U) 평균거리: 잔기 i–j 사이의 RMS Cα–Cα 거리를 계산
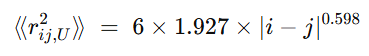
즉, 서열 간격 |i−j|만 알면 U에서의 대표 거리 스케일을 얻음 (루트 취해 RMS 거리)
접힘(F) 거리: ESMFold로 얻은 접힘 구조에서 같은 i–j의 실제 Cα–Cα 거리를 뽑음(접힘 상태의 대리값으로 사용)
distogram(거리 히스토그램) 라벨 구성: 거리축을 2–42 Å, 2 Å 간격(총 21 bin)으로 나눠 확률 히스토그램을 만듦(=distogram)
각 (i,j) 쌍에 대해
F에서 나온 거리 (𝑟𝑖𝑗,𝐹) 가 속한 bin에 확률 𝛼
U에서 나온 거리 (𝑟𝑖𝑗,𝑈) 가 속한 bin에 확률 1−𝛼
나머지 bin은 0
이렇게 해서 평형 혼합 분포 라벨을 만듦(그림 1B–C)
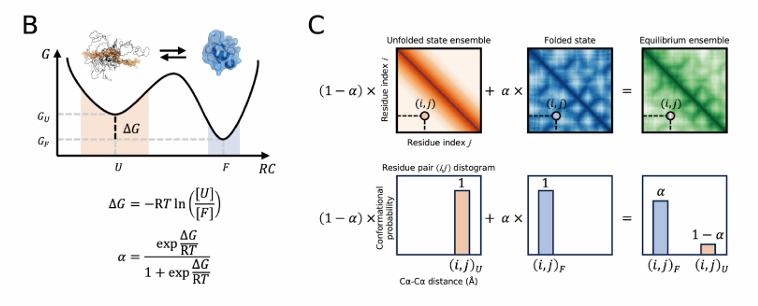
B – 에너지 경관과 α(접힘 분율)
C – 평형 distogram 구성
여기서 𝛼는 위의 두-상태 평형에서 온 접힘 분율
풀림 distogram과 접힘 distogram을 각각 (1−α), α로 가중해 합하여 평형 앙상블 distogram을 만든다. 아래쪽 막대그래프 예시에서는 특정 잔기쌍 (i,j)에 대해 왼쪽(풀림), 가운데(접힘), 오른쪽(평형) 분포가 어떻게 섞이는지 보여줌
모델 학습: 네트워크는 이 평형 distogram(i,j 전쌍의 분포)과 ΔG를 동시에 맞추도록 학습
즉, “ΔG가 크면 접힘 분율 𝛼가 커져서 분포가 F에 가깝고, 작으면 U에 가깝다”는 물리 제약을 라벨에 직접 박아 주는 것
풀림 상태를 수많은 스냅샷으로 샘플링하지 않고, Flory 법칙이 주는 평균 거리 스케일로 “U의 대표 거리”를 잡아줌
그리고 ΔG로 정해지는 가중치 𝛼를 써서 F-거리와 U-거리를 혼합한 확률분포(=distogram)를 만들면, 모델이 ΔG ↔ 거리분포의 물리관계를 함께 배우게 됨
작은 예시
서열 간격∣i−j∣=10이면, 위 식으로 U의 RMS 거리는 약 6.8 Å. 접힘 구조에서 같은 쌍의 F-거리가 예를 들어 5.0 Å라고 하자.
bin 폭이 2 Å이므로
5.0 Å → [4,6) bin,
6.8 Å → [6,8) bin.
ΔG=3 kcal/mol(25 °C)면
α≈0.994이므로, [4,6) bin에 0.994, [6,8) bin에 0.006을 주는 라벨이 된다. (다른 bin은 0.)
이렇게 (i,j) 모든 쌍에 대해 라벨을 만들어 학습한다.
✅ IFUM 프레임워크(개요)
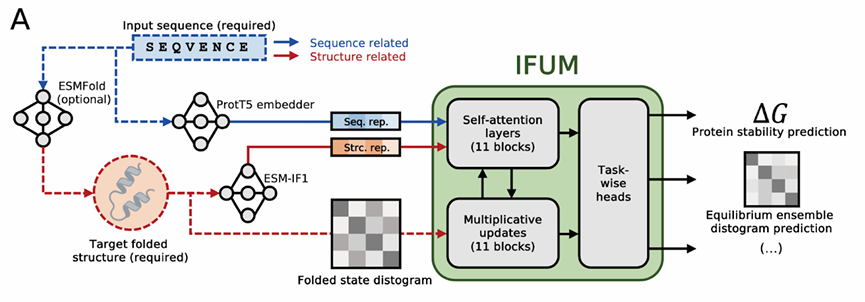
입력:
(1) 서열 임베딩: ProtT5(파란 실선)
(2) 접힌 상태 구조 임베딩: ESM-IF1(빨간 실선, ESMFold 등으로 예측한 구조를 입력으로 사용)
(3) 접힌 상태 distogram: 잔기쌍 Cα-Cα 거리를 구간화한 확률 분포, 평균 Cα-Cα 거리(빨간 점선, ESMFold 구조에서 계산된 평균 Cα-Cα 거리를 binning)
접힌 상태 distogram은 2–42 Å 범위, 21개 bin의 원-핫 인코딩 형태로 제공
(4) 풀린 상태 입력은 별도로 주지 않음(Flory가 잔기간격 |i−j|만 쓰므로).
모델 본체: AlphaFold2 Evoformer에서 영감받은 트랜스포머 모듈이 위 임베딩과 distogram을 공동 업데이트
메인 모듈은 AlphaFold2 Evoformer를 변형한 것으로, triangle attention은 메모리 이슈로 제거했지만 triangle multiplicative update는 유지
모든 self-attention은 differential self-attention으로 대체했고, pair bias와 outer product mean도 원 Evoformer처럼 씀
이런 메인 블록을 11개 쌓음
헤드들:
(i) ΔG 회귀 헤드: 잔기별 ΔG를 내고 이를 합해 단백질 총 ΔG 산출
(ii) 평형 앙상블 distogram 헤드: 위의 혼합 규칙을 따르는 확률분포를 예측
→ 두 과제를 함께 학습하면, ΔG만 단독으로 학습할 때보다 ΔG 예측이 유의하게 향상
(iii) 서열 복원 보조 헤드
평형 앙상블(Fig 1b, c): ΔG로부터 구한 접힌 상태 분율 α를 사용, 접힌/풀린 distogram을 α와 (1−α)로 가중 평균해 평형 distogram을 구성
안정한 서열은 𝛼가 커서 접힌 distogram과 비슷한 예측 분포(선명하고 질서정연)가 나오고, 불안정한 서열은 혼합(distogram이 퍼짐)에 가깝게 나옴
실제로 예측 distogram 패턴이 ΔG와 정합적임을 보임
α (folded population): 주어진 조건에서 접힌 상태의 평형 점유율
ΔG가 클수록 α↑
가중치로 써서 접힘/풀림 앙상블을 합성
데이터 구축(학습 라벨과 테스트 세트)
ΔG 라벨
(1) Mega-scale: cDNA 디스플레이 프로테오라이시스에서 유도한 ΔG 라벨(소형 단백질)
(2) DisProt: 무질서 단백질은 “ΔG < 0.5 kcal/mol”로 설정
(3) distogram 라벨: 접힘(ESMFold 거리)·풀림(Flory 평균거리)을 볼츠만 비율로 원-핫 distogram에 인코딩
볼츠만 비율(Boltzmann ratio): 열평형에서 어떤 상태 𝑠의 점유확률은 볼츠만 가중치에 비례
-> 즉, 자유에너지 차이로부터 상태 점유비를 주는 지수관계
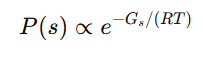
Gs는 그 상태의 깁스 자유에너지
두 상태(접힘 F, 풀림 U)의 점유비(=볼츠만 비율)
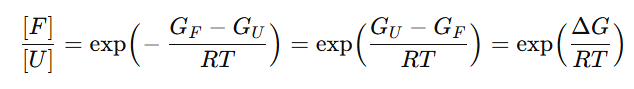
이 논문은
Δ𝐺≡𝐺_unfolded−𝐺_folded 로 두고,
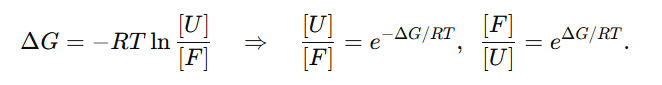
이걸 접힘 분율 𝛼로 쓰면
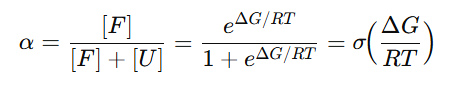
논문은 이 𝛼를 접힘/풀림 거리분포를 섞는 가중치로 사용해 “평형 distogram” 라벨을 만든 뒤, 그 distogram까지 함께 예측하도록 학습시켜 ΔG 예측을 강화
평형 distogram 라벨: 접힘 평균거리(예측 구조로부터)와 풀림 평균거리(Flory)를 원-핫 distogram으로 만들고, 볼츠만 비율(패널 B의 α,1−α)로 가중하여 최종 라벨을 구성.
이렇게 하면 해당 서열이 변성(풀림) 구조를 어느 정도 수용하는지를 학습
훈련·검증 분할
학습: Mega-scale 소단백질 648,650개 + DisProt 3,219개
평가(오로지 테스트 전용):
(i) CATH 유래 자연 단백질 6,007개(최대 869 aa),
(ii) IFN-λ·IL-10·UGT76G1 엔지니어링 변이 26개(Tm 보유),
(iii) 문헌 야생형 40개(ΔG),
(iv) de novo 설계 413개(발현 데이터 포함). Mega-scale 학습셋과 30% 이상 동일성 없음으로 누수 차단
“접힌 구조=ESMFold” 근사의 한계 인지
구조예측 AI가 파괴적 변이에 둔감하여 WT와 거의 같은 구조를 내놓는 경우가 있음
그래서 IFUM은 ESMFold 구조를 ‘목표 접힘 상태’의 대용으로 쓰되, 이 한계를 인정
(그 때문에 풀린 상태 앙상블을 함께 넣는 설계가 필요한 셈)
풀린 상태를 무시하면 ΔG의 절반을 버리는 셈이므로, Flory로 평균화한 U-distogram을 접힘 분율 α(=sigmoid(ΔG/RT))로 F-distogram과 혼합해 평형 분포를 라벨로 만들고, 모델이 그 분포 자체와 ΔG를 함께 맞추도록 학습
이 물리 일관성이 ΔG 회귀의 일반화를 크게 밀어줌
💡 Result
✅ IFUM performance on Mega-scale and other datasets
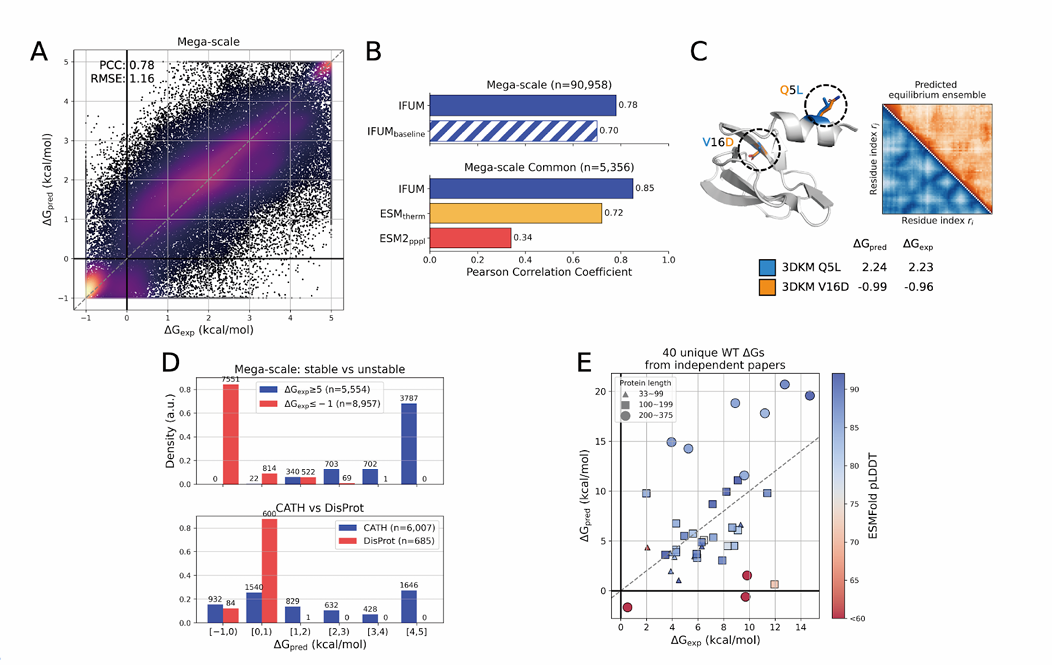
A – Mega-scale 테스트 세트 정밀도
ΔG_pred vs ΔG_exp 산점도(색 농도=점 밀도).
ΔG는 실험의 동적범위를 따라 [−1, 5] kcal/mol로 클램프.
이 설정에서 RMSE=1.16 kcal/mol, PCC=0.78을 얻음
B – 비교(위: 내부 baseline, 아래: 외부 모델)
Unfolded 앙상블 모델링을 뺀 IFUM-baseline과 비교 시 PCC가 0.78→0.70로 하락, RMSE는 1.16→1.39로 악화
즉 풀린 상태 앙상블 예측을 함께 학습해야 ΔG가 좋아짐
ESMtherm(ΔG용 파인튜닝 모델), ESM2 pseudo-perplexity와 공통 테스트(Mega-scale Common, 5,356개)에서 비교: IFUM PCC=0.85, ESMtherm 0.72, ESM2 0.34. 공정 비교를 위해 두 모델의 테스트셋 겹치는 서열만 사용
IFUM·ESMtherm 양쪽 테스트셋에 겹치는 서열만 추려 5,356개로 구성. 여기서 IFUM PCC 0.85로 ESMtherm(0.72)·ESM2(0.34)보다 높았음
C – 구조/분포 예시
HECTD1 CPH 도메인(3DKM) 돌연변이 Q5L vs V16D: ESMFold 구조 오버레이와 함께 두 변이의 ΔG_pred/ΔG_exp, 그리고 예측 평형 distogram(Q5L 파란색, V16D 주황색)을 나란히 제시.
D – 부분집합 히스토그램
Mega-scale 테스트에서 매우 안정(ΔG_exp ≥5) vs 매우 불안정(ΔG_exp ≤−1) 집합의 ΔG_pred 분포를 비교(위)
CATH(접힘 도메인) vs DisProt(무질서) 집합의 ΔG_pred 분포도 비교(아래).
축 표기에서 괄호()=배타, 대괄호[]=포함 경계를 명시.
통계적으로도 안정/불안정 구분이 유의(Welch t-test p ≪ 0.001).
또한 CATH/DisProt의 반-라벨 데이터에서도 같은 구분을 실험
E – 문헌 WT 40종 산점도
ΔG_pred vs ΔG_exp 산점도에 점 크기=길이, 점 색=ESMFold pLDDT로 시각화, 대각선은 완전일치선
✅ “Unfolded ensemble”을 함께 예측하면 ΔG가 왜 더 잘 맞나?
‘풀린 상태 앙상블(distogram) 예측’이라는 보조 목표를 빼고 단순화한 베이스라인(IFUM_baseline)을 학습해 보니, PCC가 0.78→0.70로 하락, RMSE도 1.16→1.39 kcal/mol로 악화
(ΔG 레이블은 [−1,5] kcal/mol 클램프 기준)
즉, 풀린 상태 앙상블을 함께 학습해야 ΔG 회귀가 좋아진다는 증거
왜 그런가? (모델이 배우는 신호)
모델이 예측한 “평형 distogram”(접힘/풀림을 α, 1−α로 섞은 분포)을 보면 안정 단백질은 접힘 분포에 가까운 ‘질서정연한’ 모양, 불안정 단백질은 접힘+풀림이 섞인 ‘퍼진’ 모양이 나와서 ΔG 예측과 정합성이 높음
즉, 물리적 제약(볼츠만 가중 혼합)을 따르는 보조 과제가 본 과제(ΔG 회귀)의 일반화를 끌어올린 것
추가 어블레이션(아키텍처 요소)
Evoformer류의 triangle multiplicative updates를 제거하거나, 저자들이 쓴 differential self-attention을 일반 self-attention으로 모두 교체하면 성능이 유의하게 떨어짐
그런데 평형 distogram 예측 목표를 뺀 경우에도 성능 하락 폭이 맞먹을 정도였다고 보고
결론: ΔG 추정에서 ‘평형 앙상블 모델링’의 기여가, 최신 아키텍처 트릭들에 버금갈 만큼 크다
✅ 다양한 단백질에 폭넓게 적용 가능
(i) Mega-scale 테스트에서의 판별력
잘 통제된 조건(PBS, pH 7.4, 298 K)에서 수집된 Mega-scale 테스트셋 내부에서, 매우 불안정(ΔG_exp ≤ −1) vs 매우 안정(ΔG_exp ≥ 5)을 유의하게 구분함(Welch’s t-test p≪0.001).
즉, 절대값 수준에서 안정/불안정 클래스를 잘 갈라낸다는 뜻
(ii) 자연 단백질(반(半)라벨 데이터)로의 일반화
CATH에서 뽑은 구조화 도메인 6,007개와 DisProt의 무질서 단백질 685개에 대해 ΔG_pred의 분포가 서로 다름을 확인했고, 통계적으로도 p≪0.001.
즉, 자연 단백질 콘텍스트에서도 안정성 정도 차이를 반영하는 신호를 낸다.
(iii) 낮은 ΔG_pred를 보이는 CATH 도메인의 해석(주의점)
용매에 노출된 소수성 잔기가 많은 도메인들(예: 의무적 올리고머 인터페이스, 막단백질의 막 삽입 영역)은 고립된 도메인만 떼어 보면 물 안에서 스스로 ‘불안정’처럼 보일 수 있음
이를 정량하려고 Rosetta SapScoreMetric으로 SAP(Spatial Aggregation Propensity)를 계산했더니, 일반 수용성 단백질의 평균 per-residue SAP ≈ 0.4–0.5와 달리, 이런 ‘in-water self-unstable’ 도메인들은 평균 SAP가 유의하게 높았음(평균 1.08 근처)
→ 맥락(다중체/막 환경)이 빠진 도메인에 IFUM을 적용할 때는 해석에 주의 필요
(iv) ‘품질 좋은 구조’가 들어오면 더 정확
실험 ΔG가 있는 40개 WT(길이 33–375 aa)로 테스트하면 전체 PCC=0.47로, Mega-scale에 비해 낮음
그런데 입력으로 쓴 접힘 구조의 pLDDT_ESMFold가 높을수록 상관이 올라가서, pLDDT>80(34개)이면 PCC=0.61, pLDDT>90(16개)이면 PCC=0.73.
→ 입력 접힘 구조 품질(신뢰도)가 예측 정확도에 직접 영향
✅ IFUM can accurately predict ΔΔG for various types of mutants
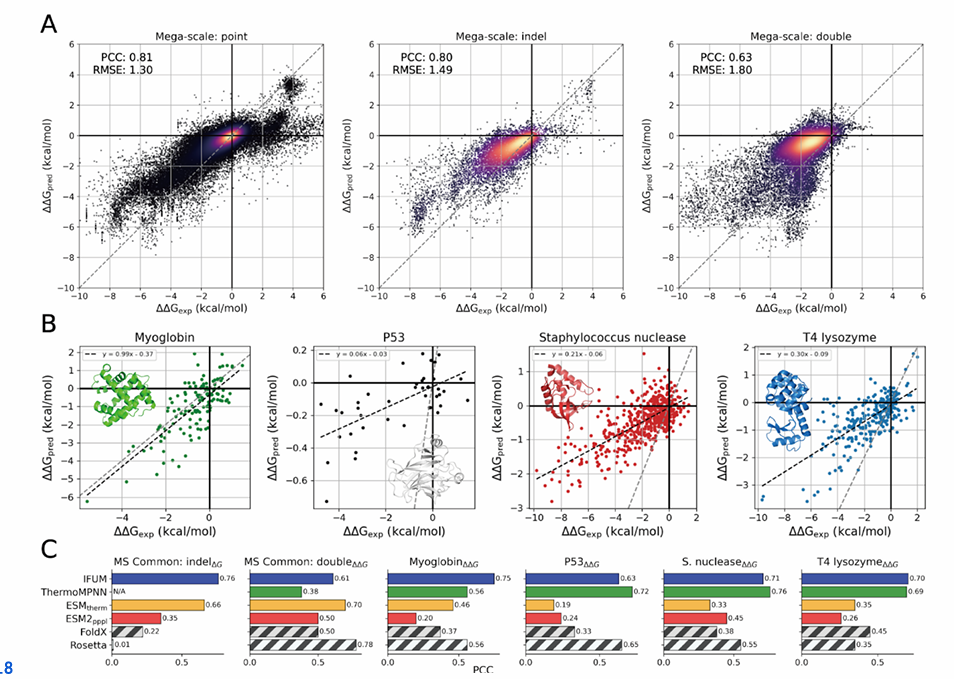
A: Mega-scale 테스트셋 안에서 변이 유형별로 ΔΔG_pred vs ΔΔG_exp 산점도
왼쪽부터 점돌연변이, 삽입/결실(indel), 이중변이
점 색 농도는 데이터 밀도(밝을수록 고밀도), 회색 점선은 완전 일치선을 뜻함
1) 변이 유형별 IFUM 단독 성능 (Mega-scale 전체 테스트셋)
IFUM의 PCC는 점돌연변이 0.81, indel 0.80, 이중변이 0.63으로 보고
즉, 단일·indel에서 특히 강하고, 이중변이에서도 유의한 상관을 보였다는 뜻
B: 4개 케이스 스터디(마이오글로빈, p53, Staphylococcal nuclease, T4 lysozyme)에 대해 ΔΔG_pred vs ΔΔG_exp 산점도
회색 점선=완전 일치, 검은 점선=회귀선
2) 케이스 스터디 4종(모두 점돌연변이 ΔΔG)
IFUM PCC: 마이오글로빈 0.75, p53 0.63, S. nuclease 0.71, T4 lysozyme 0.70.
ThermoMPNN(점돌연변이 전용)과 비교하면 각각 0.56, 0.72, 0.76, 0.69로, 일부는 IFUM이 더 좋고, 일부는 ThermoMPNN이 더 좋지만 전반적으론 비슷한 수준
ESM_therm/ESM2는 상대적으로 낮은 재현성을 보였다고 정리
C: 여러 방법(IFUM, ThermoMPNN, ESM_therm, ESM2_ppl, FoldX, Rosetta)의 PCC(상관계수) 막대 비교
비교는 MS Common indel(ΔG), MS Common double(ΔΔG), 네 케이스 스터디(ΔΔG)에 대해 수행
3) 공정 비교 세팅(MS Common)과 타 방법 대비
비교의 공정성을 위해, IFUM Mega-scale 테스트셋과 ESM_therm 테스트셋의 교집합 5,356개로 MS Common을 만들고 여기에 여러 방법을 같이 돌림
Indel (MS Common)
ThermoMPNN은 indel을 지원하지 않아서 비교 불가.
이 조건에서 IFUM은 PCC 0.76으로, ESM_therm 0.66 / ESM2 0.35 / FoldX 0.22 / Rosetta 0.01보다 앞섬
(참고: ThermoMPNN은 점돌연변이 전용이라 WT·변이 서열 길이가 달라지는 indel을 처리할 수 없다고 명시돼 있음)
이중변이 (MS Common)
IFUM 0.61, ThermoMPNN-D 0.38, ESM_therm 0.70, ESM2 0.50, FoldX 0.50, Rosetta 0.78.
즉, ThermoMPNN-D와 ESM2/FoldX 대비 우수했고, ESM_therm·Rosetta가 더 높게 나오는 조건도 존재했음을 보여줌
중요한 포인트는 *방법 전반을 아우르는 ‘일관성’
결론: 테스트한 방법들 중에서, IFUM만이 점돌연변이→이중변이→indel까지 폭넓은 변이 유형에서 ‘지속적으로’ 작동했다는 점을 강조
내부 성능:
Mega-scale 전범위에서 점·indel·이중 모두 유의한 상관(0.81/0.80/0.63)을 달성
공정 비교(MS Common):
Indel: IFUM이 모든 비교법보다 높음(0.76)
특히 indel 비지원 모델과 전통 물리기반( FoldX/Rosetta ) 대비 강점
이중변이: IFUM 0.61로 ThermoMPNN-D/ESM2/FoldX보다 우수, ESM_therm·Rosetta가 더 높게 나오는 사례도 있음
단백질별 케이스 스터디(점변이): IFUM 0.75/0.63/0.71/0.70, 전용 모델 ThermoMPNN과 비슷한 레벨, PLM 기반 단독 지표는 상대적으로 약함
총평: 유형 불문(점·이중·indel)으로 일관되게 쓸 수 있는 단일 모델이라는 점이 IFUM의 가장 큰 장점
✅ 실제 단백질 공학 및 계산적 스크리닝에서의 IFUM 성능
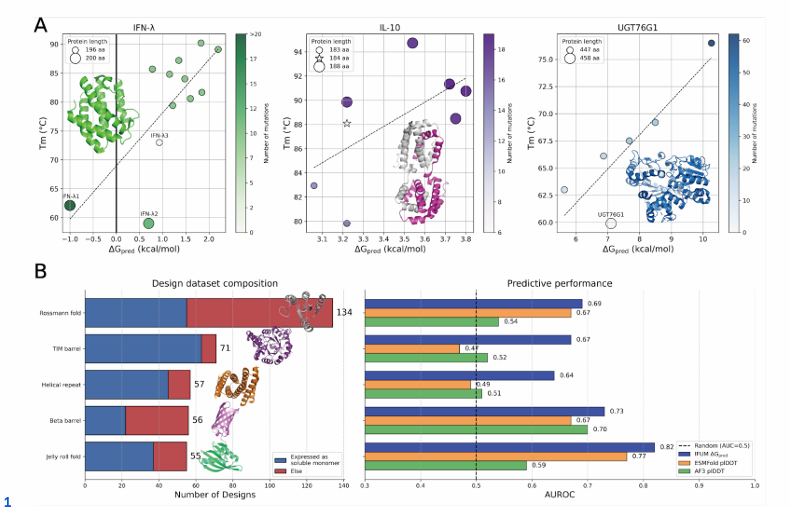
(A) IFN-λ, IL-10, UGT76G1의 서열/백본 재설계에 대해, 실험적 융해온도(T_m)와 IFUM이 예측한 ΔG(ΔG_pred)를 산점도로 나타냄
점의 크기는 단백질 길이를, 색은 각 야생형 대비 도입된 변이 개수를 뜻함
라벨이 달린 표지는 각 야생형을 가리킴
산점도에는 Rosetta FastRelaxed AF3 모델 구조를 사용했고, 점선은 선형 회귀선
점이 오른쪽 위로 갈수록(ΔG_pred↑, Tₘ↑) 안정성이 높은 변이라는 뜻.
세 타깃( IFN-λ, IL-10, UGT76G1 ) 모두에서 양의 상관이 나옴
( PCC=0.75/0.62/0.87 )
표본이 작아 p값은 각각 0.005/0.104/0.023으로 보고됨
(IL-10은 표본 한계로 유의성 약함).
같은 조건에서 AF3의 plDDT는 Tₘ와 덜 일관했고, IL-10/UGT76G1에서는 음의 상관까지 나왔음
즉, “구조 신뢰도”만으로 안정화 설계를 고르기 어렵고, IFUM ΔG_pred가 더 직접적
세 타깃 모두 다중 치환과 길이 변화(삽입/삭제)가 섞여 있는데, 이런 난이도 높은 안정화 문제에서 IFUM이 범용적으로 통한다는 걸 시각적으로 확인시켜 줌
(B) 설계 단백질의 in silico 스크리닝 성능
왼쪽: 5가지 단백질 폴드에 대한 설계 데이터셋 구성을 보여주며, 실험으로 대장균에서 가용성 단량체로 발현된 설계(파랑)와 그렇지 않은 설계(빨강)의 수를 나타냄
오른쪽: 위 과제를 “발현 성공/실패”로 이진 분류했을 때의 AUROC. IFUM ΔG_pred(파랑) vs ESMFold/AF3 plDDT(주황/초록) 비교
IFUM의 ΔG_pred(파랑)를 ESMFold(주황)와 AF3(초록)의 plDDT와 비교
모든 스캐폴드에서 IFUM ΔG_pred가 plDDT보다 AUROC가 높음
→ “발현될 가능성이 높은 설계”를 더 잘 가려냄.
✅ 실용적 응용 1. 단백질 안정화 공학
단백질 공학에서는 길이 변형이나 다중 치환을 흔히 도입
안정화를 위해 루프를 잘라내기도 하고, 새 기능을 넣으려면 오히려 길게 만들기도 함
구조예측 신뢰도 지표가 이런 작업의 가이드(필터링 등)로 널리 쓰이지만, 양적 기여는 불명확
여기서는 IFUM이 이런 지표의 좋은 대안이 될 수 있음을 보임
이 비교에서 네트워크의 접힌 상태 입력 구조는 Rosetta FastRelaxed AF3 모델을 사용
세 가지 사례를 테스트
(i) IFN-λ3: 트롬빈 절단부위를 담는 유연 루프를 도려내고 구조적으로 인페인팅한 α-나선으로 대체해 인접 소수성 패치를 가렸고, 프로테아제 분해 저항성을 높임
야생형 IFN-λ1/2/3과 9종 변이체의 T_m을 열전이 분석으로 측정
(ii) IL-10: 단량체형 IL-10 변이 8종에서, 야생형 대비 일부 잔기를 삭제하고(11개), 변이에 따라 15개 또는 20개를 삽입(각각 2종, 5종)하여 전체 4–9개 잔기 길이가 늘음
비교를 위해 GS 링크가 들어간 단량체형 IL-10M1도 포함
(iii) UGT76G1: 이전 보고의 야생형과 5종 재설계 변이에 대해 T_m을 정리했으며, 원래 길이(458 aa) 대비 14–62개의 치환(그중 11개 삭제)이 도입
각 서열의 최종 변이 수는 야생형과의 정렬로 다시 셈
결과적으로 세 그룹 모두에서 예측 ΔG_pred와 실험 T_m 사이에 좋은 양의 상관을 보임
즉, 후보 변이들을 ΔG_pred 기준으로 우선순위를 매기고, 그래프처럼 Tₘ 대체 지표로 삼음
동일 백본·조건에서라면 ΔG_pred가 클수록 Tₘ가 오를 확률이 높음
-> 각각 IFN-λ: PCC 0.75, IL-10: 0.62, UGT76G1: 0.87; 표본 수가 작아 해석에 주의: p=0.005, 0.104, 0.023
대조적으로 AF3의 신뢰도 지표(plDDT)는 IL-10과 UGT76G1에서는 오히려 음의 상관을 보임 (Fig. S8)
이는 길이 변화와 다중 치환이 동시에 일어나는 까다로운 안정화 문제에서 IFUM이 AF3 지표보다 넓게 일반화됨을 시사
✅ 실용적 응용 2. de novo 설계 단백질의 가용성 단량체 발현 스크리닝
계산 설계의 최종 단계에서 구조예측 모델로 후보 서열을 우선순위화하기도 함
여기서는 E. coli에서 가용성 단량체로 발현되는 설계를 골라내는 능력을 IFUM, ESMFold, AF3 사이에서 비교
IFUM의 ΔG_pred로 필터링하면 plDDT 기반 필터(ESMFold/AF3)보다 일관되게 더 나은 구분 성능(AUROC)을 보임
이 결과는 Rossmann, TIM 배럴, helical repeat, β-barrel, jelly-roll의 다섯 가지 스캐폴드 전반에서 확인되었고, 학습 데이터와의 서열 유사도가 0.30을 넘는 설계는 사전에 제외
💡 Discussion
✅ 왜 ‘unfolded 상태 앙상블’을 명시적으로 넣으면 좋아지나?
ΔG는 “접힌 상태와 미접힌 상태의 자유에너지 차이” 그 자체라서, 두 상태를 함께 다루는 게 원리적으로 맞음
IFUM은 실제로 그렇게 했고, 정확도가 올라갔다고 보고
증거는 두 가지:
1) 단일 접힌 구조(보통 WT 구조)에만 의존하는 기존 ΔΔG 예측기들은 복잡한 변이 유형(예: 다중변이, 인델 등)을 포착하는 데 한계가 드러났음(IFUM의 벤치마크 비교에서 확인)
2) IFUM에서 평형 앙상블 예측 목표를 빼면 성능이 크게 떨어지는데, 그 폭이 네트워크의 중요한 구성요소(예: 삼각 곱 업데이트 등)를 제거했을 때의 하락폭과 맞먹을 정도
(= 앙상블 모델링 자체가 ‘알고리즘적 핵심 기여’라는 뜻)
✅ 대규모 사전학습(transfer learning)이 과적합을 줄여서 견고성을 준다
IFUM은 ProtT5(서열 임베딩)와 ESM-IF1(구조 임베딩)에서 뽑은 표현, 그리고 구조 입력을 결합
ProtT5는 BFD 21억 + UniRef50 4,500만 서열로, ESM-IF1은 AF2 예측구조 1,200만 개로 학습되어 있음
이런 거대한 사전학습 지식 덕분에 IFUM은 소규모 태스크 미세조정에서 흔한 과적합을 완화( ThermoMPNN도 같은 전략)
반대로 ESMtherm처럼 단일 데이터셋에 전체 모델을 파인튜닝하면 과적합 문제가 컸다고 지적
✅ 입력 ‘접힌 상태’ 구조의 품질이 예측력에 영향을 준다
자연 단백질(최대 ~375 aa)에서, plDDT>90의 AF3 모델을 입력으로 쓴 서브셋은 실험치와의 상관이 크게 향상(그림 2E).
엔지니어드 단백질들에서도 Rosetta FastRelax된 AF3 모델 → ESMFold 모델로 바꾸면, Tm–ΔGpred 상관이 뚝 떨어짐(그림 S9).
네트워크가 “입력 구조 표현이 실제 접힌 상태를 제대로 반영한다”는 가정 위에 있으니, 구조 품질이 낮으면 예측이 흔들린다는 논리
추가 분석(그림 S10)에서도, 품질 낮은 ESMFold 구조를 쓰면 WT 단백질에 대해 말도 안 되게 낮은 ΔG를 내다가, AF3 구조로 바꾸면 훨씬 그럴듯해진다고 보고
결론: 신뢰도 높은 구조를 입력으로 줄수록 IFUM이 안정적이다
✅ Potential application: 잔기 단위(Per-residue) 점수로 설계 타깃 고르기
IFUM의 단백질 ΔGpred는 잔기별 기여도의 합으로 나옴
이 잔기 분해값을 이용해 “우선 수정할 잔기”를 고를 수 있다는 아이디어
이를 시험하려고 de novo 설계 단백질에 대해 ProteinMPNN으로 두 전략 비교:
전체 리디자인 vs IFUM이 ‘음수(=불안정화)’ 기여로 찍은 잔기만 선택적 리디자인
결과: 선택적 리디자인 쪽이 원 설계 대비 ΔGpred 개선, 반면 전체 리디자인은 개선이 없었음(그림 S11).
다만 이건 모델-내 자기일관성의 산물일 수 있으므로 실험 검증이 필요
✅ Limitations of IFUM (제약 및 주의점)
학습 라벨의 범위/길이 제약
주 학습세트(Mega-scale)는 ΔG 라벨 동적범위가 [–1, 5] kcal/mol이고, 단백질 길이가 최대 80 aa 수준
이 범위를 넘어서는 매우 안정적 단백질이나 큰 단백질로 외삽할 때 정확도에 제약이 생길 수 있음
✅ 단순화된 ‘두 상태’ 모델 가정
IFUM은 이산적/단순화된 두-상태 앙상블(접힘/미접힘) 표현을 씀
실제는 연속적이고 복잡한 준위들이 존재하므로, 특히 대형/다영역 단백질의 접힘 풍경을 충분히 포착하지 못할 수 있음(그럼에도 두-상태 근사는 널리 쓰이긴 함)
✅ 막단백질/의존성 올리고머 미적용
주로 수용성 단량체 위주 데이터로 학습되어 막단백질이나 의존적 올리고머에는 바로 적용되지 않음
✅ 사전학습 의존성
대규모 사전학습의 이점(과적합 완화)과 동시에, 파운데이션 모델·데이터의 품질/범위에 성능이 묶여 있음
즉, 사전학습 없이 단독으로 어느 정도까지 통용될지는 더 입증이 필요
💡 Concluding remark
IFUM은 다양한 테스트셋에서 강한 성능을 보였고, 특히 다중변이/길이변화가 있는 엔지니어드 단백질에서 ΔGpred–Tm 상관으로 실전 유틸리티를 증명
따라서 de novo 디자인이나 엔지니어링 워크플로우에서 plDDT/PAE 같은 표준 필터를 보완하는 안정성 필터로 쓸 수 있음
💡 Methods
✅ Dataset preparation (데이터셋 준비)
1) Mega-scale (학습 라벨)
ΔG 라벨이 신뢰할 수 없음(dG_ML=“—”)으로 표시된 항목 제거.
scrambled 서열 중 ΔG가 0.5 kcal/mol 초과인 건 응집(aggregation) 산물일 수 있어 제외.
최종 829,927 서열 남겼고, 해당 deltaG 값을 훈련 라벨로 사용.
2) DisProt (미접힘 특징 학습용)
70 aa 미만 서열만 모아 IDP 풍부하게 큐레이션 → 4,705 서열.
라벨 ΔG는 없지만 불안정/무질서 단백질 DB라서, 훈련 중 이들에 대해 ΔG<0.5 kcal/mol을 예측하도록 손실을 수정해 모델이 미접힘/불안정 특징을 학습하도록 유도.
3) CATH (대규모 테스트)
비표준/미확정 아미노산, 세그먼트 3개 이상인 서열 제거
→ ESMFold 예측 시 CUDA OOM 회피 위해 900 aa 미만만 채택.
Mega-scale 테스트와 서열 유사도 0.30 초과 항목 제거
→ 6,007 서열 테스트 세트. SAP 점수는 Cao et al. 프로토콜로 계산.
4) “Literature” (순수 실험 WT ΔG)
S669 데이터셋의 원 논문 92편에서 WT 단백질 ΔG와 서열을 수집.
실험 pH 7.0–7.5만 포함, 동종올리고머화 표기가 있는 서열 제외(유니프롯 주석 기반), Mega-scale train과 유사도 0.30 초과인 서열 제거 → 40개 WT 테스트 전용 세트.
5) 추가 테스트 세트
ThermoMPNN이 쓴 네 가지 case-study(p53, myoglobin, S. nuclease, T4 lysozyme)도 동일하게 test-only로 사용.
UGT76G1 WT 및 7개 리디자인 서열의 Tm(Go et al.) 수집.
다섯 가지 접힘형(fold)에서 de novo 413개 서열과 발현 데이터 수집.
✅ Design workflow of IFN-λ3 & IL-10 (설계 워크플로)
RFdiffusion으로 두 WT의 루프를 안정화 지향 재설계하고, 생성 모델 중 α-helical linker 포함 구조를 DSSP로 골라 백본으로 채택
선택한 백본에 ProteinMPNN으로 서열을 생성하고, 설계 대상이 아닌 구간은 고정.
최종 서열의 Tm 측정을 수행했고, 시퀀스는 GitHub에 공개 예정
(참고: IFN-λ3/IL-10/UGT76G1 각 설계·엔지니어링 결과와 Tm/ΔG 예측 상관은 본문/표 보조자료에 상세히 보고됨.)
✅ Dataset splitting (분할) & Ablation (소거 실험)
MMseqs2 easy-cluster로 Mega-scale과 DisProt을 최소 서열유사도 0.30 기준 군집화.
각 클러스터를 나눠 train:val/test = 8:2, 이후 val과 test를 1:1로 랜덤 분할.
Mega-scale Common: IFUM의 Mega-scale test와 ESMtherm 의 test-only 도메인이 공통으로 갖는 5,356 서열을 뽑아 두 모델 비교에 사용
(Fig. 2D, 3D).
Ablation: 동일 데이터/동일 하이퍼파라미터로 여러 변형을 학습, 검증
𝑅2가 최고인 모델을 대표로 택하고 동일 테스트 세트로 지표를 계산.
✅ Other predictors (비교용 기존 방법)
1) ThermoMPNN: ProteinMPNN을 전이학습한 GNN.
점변이 전용(서열 길이 동일 필요)이라 indel 효과는 직접 예측 못함.
본 비교에서는 단일 변이는 ThermoMPNN, 이중 변이는 ThermoMPNN-D를 사용.
2) ESM2 zero-shot: 마스크 예측 확률의 로그를 전 잔기에 합산한 pseudo-log-likelihood(plll)로 점수화 (Eq. 7)
본 연구는 esm2_t36_3B_UR50D를 사용
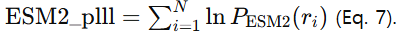
3) ESMtherm: ESM2를 Mega-scale로 파인튜닝해 ΔG를 예측하는 PLM.
공개 스크립트로 점수 생성.
4) FoldX 5.0: 에너지 함수 기반으로 ΔG/ΔΔG 계산(예측 구조 사용), indel 포함.
5) Rosetta: 물리/경험 포텐셜 기반, FastRelax와 Cartesian ddG 프로토콜(ref2015)로 ΔG/ΔΔG 계산, indel 포함.