들어가며
샘플 단위의 Gene set enrichment analysis (GSEA) 를 위해 많이 쓰이는 Gene Set Variation Analysis (GSVA; BMC Bioinformatics, 2013) 알고리즘을 리뷰하고자 한다.
2024/05/29 및 구글 스콜라 기준, 해당 논문은 8,887회 피인용 되었다.
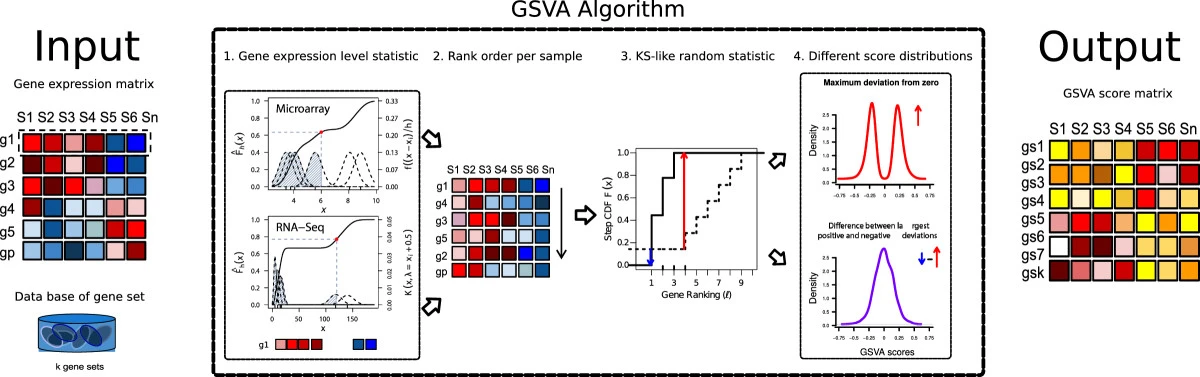
본문을 보면 몇몇 수식으로 인해 어려워 보일 수도 있지만, 상당히 간단하고 직관적인 알고리즘이다. 본 게시글에선 수식은 최대한 배제하고 단순화하여 직관적으로 설명하고자 한다.
Gene set analysis (GSA)
GSVA를 설명하기 앞서, Gene set analysis 에 대해 간략히 설명하고 넘어가고자 한다.
Gene set analysis (GSA) 는 말 그대로 유전자 집합에 대한 분석이다.
DEG 분석의 경우, 각 유전자 하나 하나의 유의도와 차이의 크기를 정량화하여 보고자 했다.
반면, GSA는 두 그룹 간 차이나는 유전자들의 대다수가 어떤 카테고리에 들어가 있는지, 집합의 단위로 보고자 한다. 혹은 반대로, 특정 카테고리에 묶이는 유전자들이 두 그룹 간에 차이가 나는지 볼 수도 있다.
내가 디자인한 실험에 따라 두 방식 중 하나를 택하며,
선택한 방식에 따라 써야 하는 도구나 알고리즘이 달라질 수 있다. (둘 다 쓰는 경우도 많다.)
전자의 방식에 적합한 분석은 Over-representation analysis (ORA) 혹은 Gene ontology analysis (GO analysis)이며, 후자의 방식에 적합한 방식이 지금 소개하고자 하는 GSVA이다.
왜 그런지 부연설명을 좀 더 하면,
전자의 방식(ORA, GO analysis)은 비교적 간단한 알고리즘들로 구성된다.
두 그룹 간 DEG 분석을 먼저 수행하고, DEG 목록, 유전자 리스트를 구성한다.
이 유전자 리스트에 Enriched, 혹은 Over-represented 된 유전자 집합 (카테고리)를 간단한 통계적 기법을 통해 찾는다. 즉, DEG가 100개 나오던, 10개가 나오던 내가 유의하다고 판단한 DEG들이 주로 어디에 속해있는지 판단하는 과정이다.
후자의 방식에 왜 GSVA가 적합한가는 마지막에 다시 설명하겠다.
Gene set
그렇다면, '유전자 카테고리'라고 내가 표현한 유전자 집합(?)은 무엇인가?
주로 쓰이는 유전자 카테고리, 즉 분류법에는 Gene ontology, KEGG Pathway, MSigDB, Reactome pathway 등이 있다.
이 중 Gene Ontology에 대해서만 간략히 설명하고 넘어가겠다.
Gene Ontology (GO)
Gene Ontology (GO)는 유전자와 유전자 산물의 특성을 일관된 용어로 표현하고 분류하는 데 사용되는 생물학적 어휘 체계이다. GO는 크게 세 가지 카테고리로 구성된다.
- Biological Process (BP): 유전자 산물이 관여하는 생물학적 과정. 예를 들면, "cell cycle", "apoptosis" 등이 있다.
- Molecular Function (MF): 유전자 산물의 분자 수준 활성. 예를 들면, "catalytic activity", "transporter activity" 등이 있다.
- Cellular Component (CC): 유전자 산물이 작용하는 세포 내 위치. 예를 들면, "nucleus", "cytoplasm" 등이 있다.
각 유전자는 이 세 가지 카테고리 아래 여러 GO term에 할당될 수 있다. GO는 생물학 연구에서 널리 사용되는 표준화된 어휘로, 유전자 집합 분석에 매우 유용하다.
(예시) TP53 유전자와 Gene Ontology
TP53 (or P53)은 종양 억제 유전자로 잘 알려져 있으며, 세포 주기 조절, DNA 복구, 세포 사멸 등 다양한 생물학적 과정에 관여한다. 이 유전자는 다음과 같은 GO term에 속한다.
Biological Process (BP):
- apoptotic process (GO:0006915)
- cell cycle arrest (GO:0007050)
- cellular response to DNA damage stimulus (GO:0006974)
negative regulation of cell proliferation (GO:0008285)
Molecular Function (MF):
- DNA binding (GO:0003677)
- p53 binding (GO:0002039)
- protein binding (GO:0005515)
- transcription factor activity, sequence-specific DNA binding (GO:0003700)
Cellular Component (CC):
- nucleus (GO:0005634)
- cytosol (GO:0005829)
이처럼 TP53은 세포 내에서 다양한 위치에 존재하며 (CC), DNA 결합, 전사 조절 등의 분자적 기능을 수행하고 (MF), 이를 통해 세포 주기 조절, 세포 사멸 등의 생물학적 과정에 관여한다 (BP).
만약 어떤 실험에서 두 그룹 간 TP53의 발현량 차이가 관찰되었다면, 이는 TP53이 관여하는 생물학적 과정, 즉 세포 주기, 세포 사멸 등에서 그룹 간 차이가 있을 가능성을 시사한다. 그러나 단순히 TP53의 발현량 차이만으로는 해당 과정들의 변화를 단정 짓기는 어렵다. 생물학적 과정은 보통 여러 유전자들의 복잡한 상호작용을 통해 조절되기 때문이다.
만약 GSA 분석 결과, 한 그룹에서 TP53의 발현 변화를 비롯해 'apoptotic process'에 속하는 다른 유전자들의 발현도 함께 변화한 것으로 나타난다면, 이는 해당 그룹에서 세포 사멸 과정(apoptotic process)의 활성화 또는 억제가 일어나고 있음을 보다 강력하게 시사한다고 볼 수 있다.
이렇게 개별 유전자의 발현 변화와 GSA 분석 결과를 종합하면, 두 그룹 간 관찰되는 생물학적 차이를 보다 명확하게 해석할 수 있다.
이외 유전자 집합 데이터베이스
- KEGG: KEGG는 유전자, 단백질, 화합물 등의 분자 수준 정보를 통합한 데이터베이스입니다. 특히 KEGG pathway는 대사, 신호 전달, 질병 등 다양한 생물학적 과정을 네트워크 그림 형태로 잘 나타내고 있어, 직관적이고 해석이 용이하다.
- MSigDB (Molecular Signatures Database): MSigDB는 생물학적 기능, 위치, 조절 관계 등 다양한 범주의 정보를 바탕으로 유전자 집합을 미리 정의해 놓은 데이터베이스이다.
- Reactome: Reactome은 인간 생물학에 초점을 맞춘 데이터베이스로, 생화학 반응과 신호 전달 과정을 중심으로 관련 유전자, 단백질, 복합체 등의 정보를 체계적으로 제공한다. 웹 접근성이 다른 DB에 비해 매우 높다.
GSVA 알고리즘
Overview
GSVA 알고리즘은 기본적으로 다음과 같은 절차로 이루어진다.
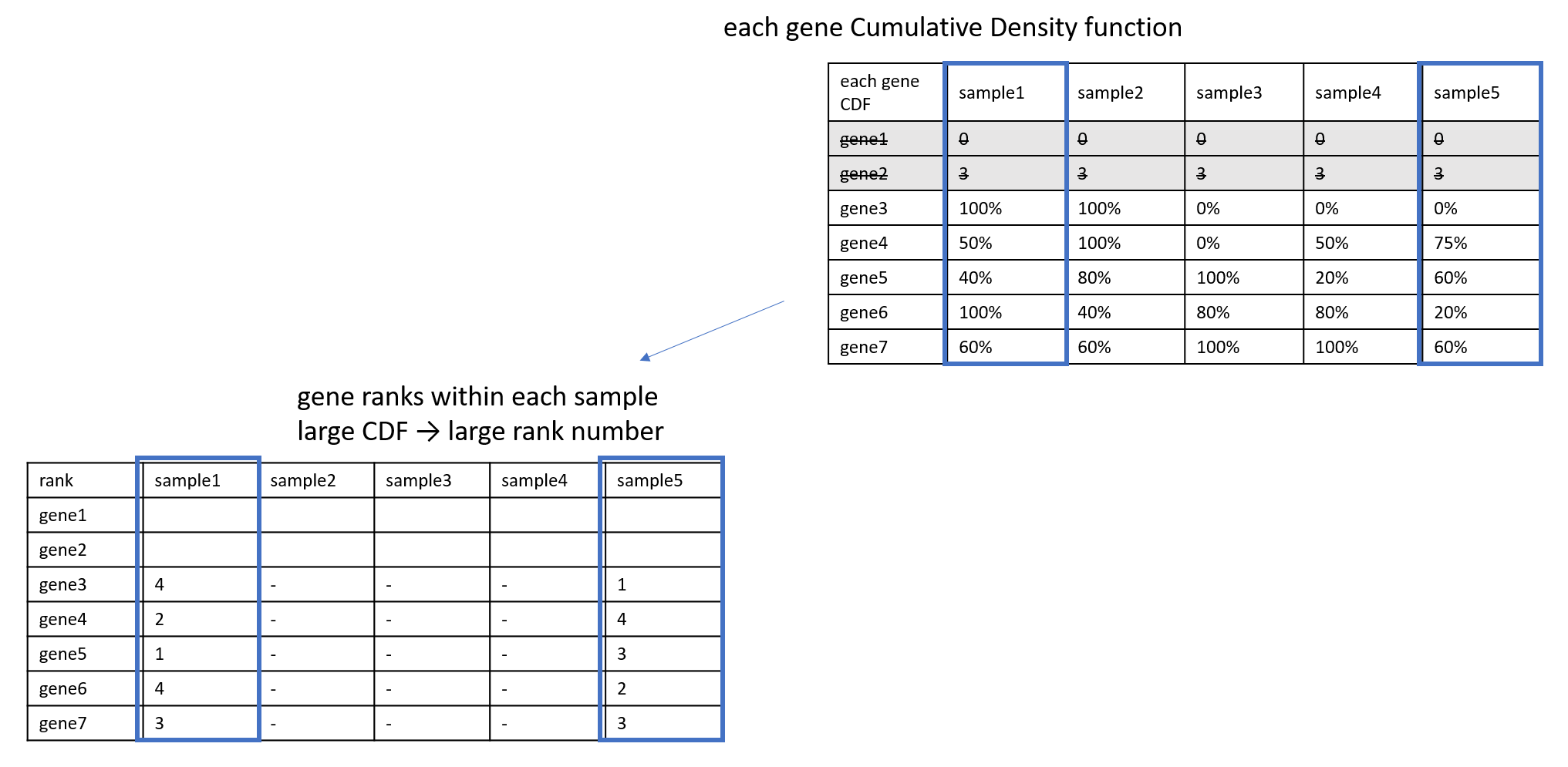
GSVA 알고리즘은 유전자 발현 데이터 (Gene expression matrix)를 입력으로 받는다. 이 행렬의 행은 샘플, 열은 유전자에 해당한다. (여기서 그룹 정보는 쓰이지 않는다.)
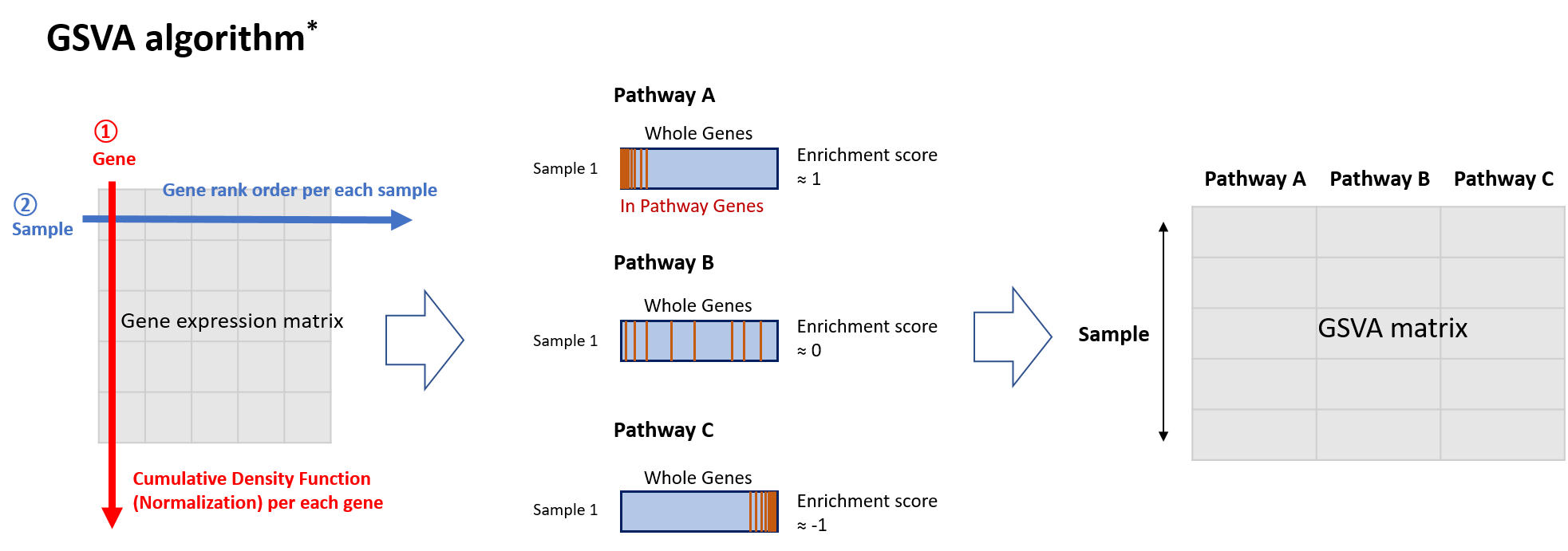
1. 유전자 기반 정규화 (그림의 붉은색 화살표, Gene 방향)
먼저, 각 유전자에 대해, 누적 밀도 함수 (Cumulative Density Function)를 계산하고 이를 기반으로 정규화(Normalization)를 수행한다. 이 과정을 통해 유전자 간 발현량 차이의 영향을 줄일 수 있다.
2. 샘플 내에서의 유전자들의 발현 순위 (그림의 파란색 화살표, Sample 방향)
다음으로, 각 샘플에 대해, 모든 유전자의 발현량 순위 (Gene rank)를 매긴다.
3. Enrichment score 계산
샘플 내에서 전체 유전자들의 발현 순위가 매겨졌을 때, 각 Pathway (혹은 Gene set)에 대한 Enrichment score를 계산할 수 있다.
위 그림의 중간에 표현한 Sample A로 3가지 시나리오를 가정해보겠다. 파란색 박스는 유전자들을 발현량 순위로 정렬했음을 표현한 것이고, 붉은색 선분은 해당 Pathway의 유전자들이 어느 순위에 위치하는지를 나타낸다.
Sample A의 전체 유전자 발현 순위를 보았을 때:
- Pathway A에 속한 유전자들의 순위가 대체로 높다면 (붉은색 선분이 상위에 몰려있음), Enrichment score는 1에 가까운 값을 가질 것이다.
- Pathway B에 속하는 유전자들이 상위와 하위에 고르게 분포해 있다면, Enrichment score는 0에 가까울 것이다.
- Pathway C에 속한 유전자들의 순위가 대체로 낮다면 (붉은색 선분이 하위에 몰려있음), Enrichment score는 -1에 가까울 것이다.
이렇게 계산된 Enrichment score는 해당 샘플에서 각 Pathway가 상대적으로 어떤 활성화 상태에 있는지를 나타내는 지표가 된다.
토이 데이터 활용 세부 알고리즘
알고리즘을 단순화하고, 단순화한 행렬을 통해 설명해보겠다.
-
행: 유전자, 열: 샘플을 나타내는 유전자 발현 행렬을 가정한다.
-
모든 샘플들이 같은 값을 지니는 유전자를 제거한다. 이는 모든 샘플에서 발현량에 차이가 없는 유전자는 분석에 도움이 되지 않기 때문이다. 예시 그림에서 회색으로 표시된 유전자들이 이에 해당한다.
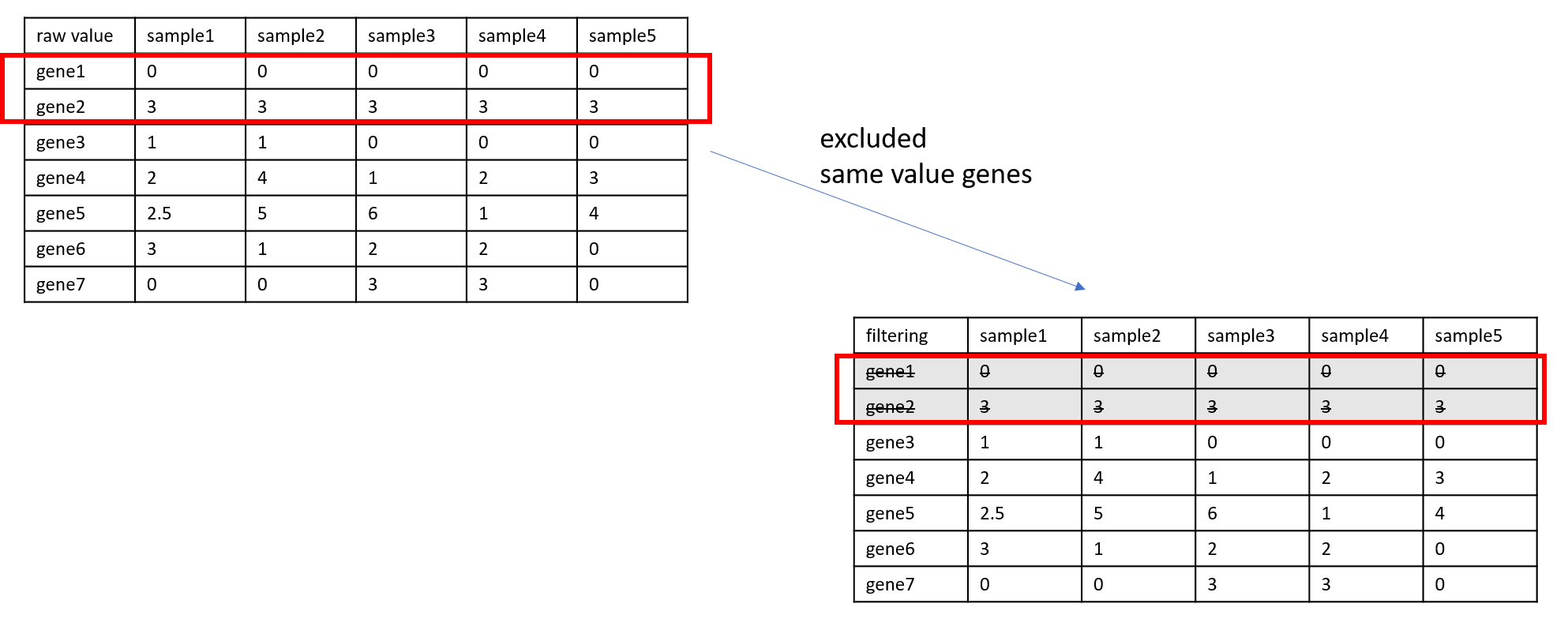
-
각 유전자별로 누적밀도함수(Cumulative Density Function, CDF)를 추정한다. 쉽게 말해서, 특정 유전자 기준으로 샘플 마다 상위 몇 %에 속하는지로 변환하는 과정이다. 최댓값은 100%, 최솟값은 0%가 된다. 이를 통해 유전자 간 발현량 차이의 영향을 줄일 수 있다. (1. 유전자 기반 정규화)

-
이제, 샘플 기준으로 넘어간다. 높은 CDF 값을 지닌 것을 높은 rank 값을 갖도록 정수화한다. (2. 샘플 내에서의 유전자들의 발현 순위)
- 여기서 Pathway 1에 gene 4, 5가 포함되어 있고, 전체 유전자는 5개 (gene3,4,5,6,7)라고 가정하겠다.
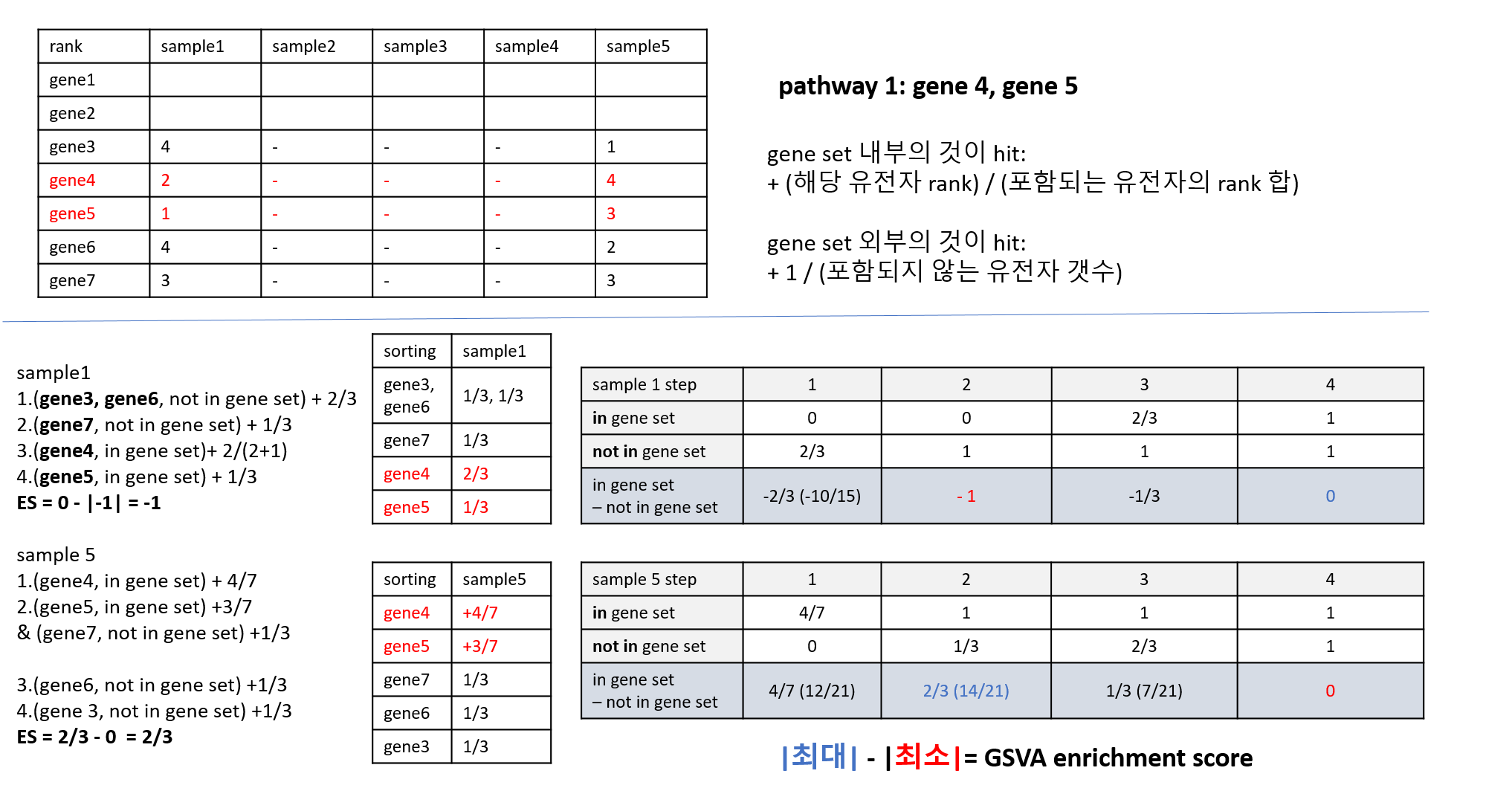
더 큰 숫자의 유전자부터 차례로 단계를 밟는데, 속하지 않는 유전자가 나올 때와, 속하는 유전자가 나올 때의 값을 별개로 계산한다.
- Pathway 1에 속하지 않는 유전자가 나오면, (not in gene set) + 1 / (Pathway 1에 속하지 않는 유전자들의 개수)
- Pathway 1에 속하는 유전자가 나오면, (in gene set) + (해당 유전자의 rank) / (Pathway 1에 속한 유전자들의 rank 합)
즉, 속하지 않는 유전자가 나올 때, 1/3 씩 not in gene set score가 가산된다.
속한 유전자가 나올때, Sample 1의 경우, in gene set에 (해당 유전자의 rank number)/3 만큼
Sample 5는, (해당 유전자의 rank number)/(7=4+3) 씩 가산된다.
in gene set score와 not in gene set score 간 차이를 각 스텝별로 계산하고, 최대와 최소 간 절댓값 차가 Enrichment score (ES)가 된다
이렇게 계산된 Enrichment score는 해당 샘플에서 특정 Pathway가 상대적으로 어떤 활성화 상태에 있는지를 나타내는 지표가 된다. Sample 1에서는 Pathway 1이 비활성화된 상태이고, Sample 5에서는 Pathway 1이 활성화된 상태임을 알 수 있다.
CDF 계산 부분/마지막 ES 계산 부분을 특히 단순화 하였다.
원 논문에서는, 마이크로어레이(microarray)의 데이터는 intensity를 나타내므로 gaussian kernel을 사용하고, RNA-seq 데이터의 경우 카운트 데이터이므로 poisson kernel을 사용하여 CDF를 추정한다.
마지막 ES 계산 부분은, KS 통계량 (Kolmogorov-Smirnov Statistics) 계산을 단순화한 것이다.
의생명 연구에서의 GSVA
GSVA는 특정 유전자 세트가 집단 간 차이가 나는지 보는 데에 적합한 방식이다. 왜냐하면, GSVA는 개별 샘플 수준에서 각 유전자 세트의 활성화 정도를 정량화하기 때문이다.
앞서 설명한 것처럼, ORA나 GO 분석은 두 그룹 간 차이나는 유전자들이 어떤 카테고리에 주로 속해있는지를 알려준다. 하지만 이는 그룹 수준에서의 분석이며, 개별 샘플에서의 유전자 세트 활성화 정도는 알 수 없다.
반면, GSVA는 각 샘플마다 유전자 세트의 활성화 점수 (Enrichment score)를 계산한다. 이를 통해 샘플 간, 그룹 간 유전자 세트의 활성화 정도를 비교할 수 있게 된다.
예를 들어, 암 조직과 정상 조직의 샘플이 있다고 하자. GSVA를 통해 각 샘플의 'Cell cycle' 관련 유전자 세트의 활성화 점수를 구할 수 있다. 만약 암 조직 샘플들에서 이 점수가 일관되게 높게 나온다면, 이는 암 조직에서 세포 주기 관련 유전자들이 전반적으로 활성화되어 있음을 시사한다.
이렇게 GSVA는 pathway나 gene set의 활성화 정도를 샘플 단위로 정량화함으로써, 집단 간 생물학적 차이를 보다 세밀하게 이해할 수 있게 해준다. 이는 단순히 두 그룹 간 차이나는 유전자 목록을 얻는 것에서 한 걸음 더 나아가, 그 차이의 생물학적 맥락을 파악하는 데 큰 도움을 준다.
하지만 GSVA는 유전자 세트별로 계산을 해야 하기 때문에, 두 집단 간 차이나는 유전자 세트를 찾기 위해서는 모든 유전자 세트에 대해 각 샘플별로 Enrichment score를 계산해야 한다. 이는 계산량이 많아질 수밖에 없음을 의미한다.
특히, 사용하는 유전자 세트의 수가 많을수록 계산 시간은 더욱 증가할 것이다. 예를 들어, MSigDB와 같이 수만 개의 유전자 세트를 포함하는 데이터베이스를 사용한다면, 모든 유전자 세트에 대해 GSVA를 수행하는 것은 상당한 계산 자원을 필요로 할 것이다.
따라서, 연구자가 이미 관심 있는 특정 pathway나 유전자 세트가 있다면, 즉 "특정 카테고리에 묶이는 유전자들 이 두 그룹 간에 차이가 나는지 보고 싶다면", 그 세트들에 대해서만 GSVA를 수행하는 것이 효율적일 수 있다.
반대로, "두 그룹 간 차이나는 유전자들이 어떤 카테고리에 주로 속해있는지 를 알고 싶다면", ORA나 GO 분석과 같은 방법이 더 적합할 수 있다. 이들은 계산량이 상대적으로 적고, 포괄적인 기능 카테고리 분석에 용이하기 때문이다.
물론, 연구의 목적에 따라 두 가지 방법을 모두 활용할 수도 있다. 중요한 것은 각 방법의 특징과 한계를 이해하고, 연구 질문에 가장 적합한 방법을 선택하는 것이다.
마치며
연구 질문과 별개로, pathway의 크기가 매우 클 경우 ES 계산이 불안정해질 수 있고, single-cell RNA-seq 데이터의 경우 zero-inflation으로 인해 순위 계산이 어려울 수 있다는 점도 고려해야 한다.
그럼에도 불구하고 GSVA는 그 심플함과 확장성 덕분에 pathway/gene set enrichment 분석에서 널리 쓰이고 있으며, 이에 기반한 새 알고리즘도 많이 제안되고 있는 알고리즘이다. 한번쯤은 꼭 들여다 볼 만한 가치가 있는 논문이라 생각한다.

그러면 GSVA enrichment score는 1과 -1 사이의 값이 나오는 건가요?