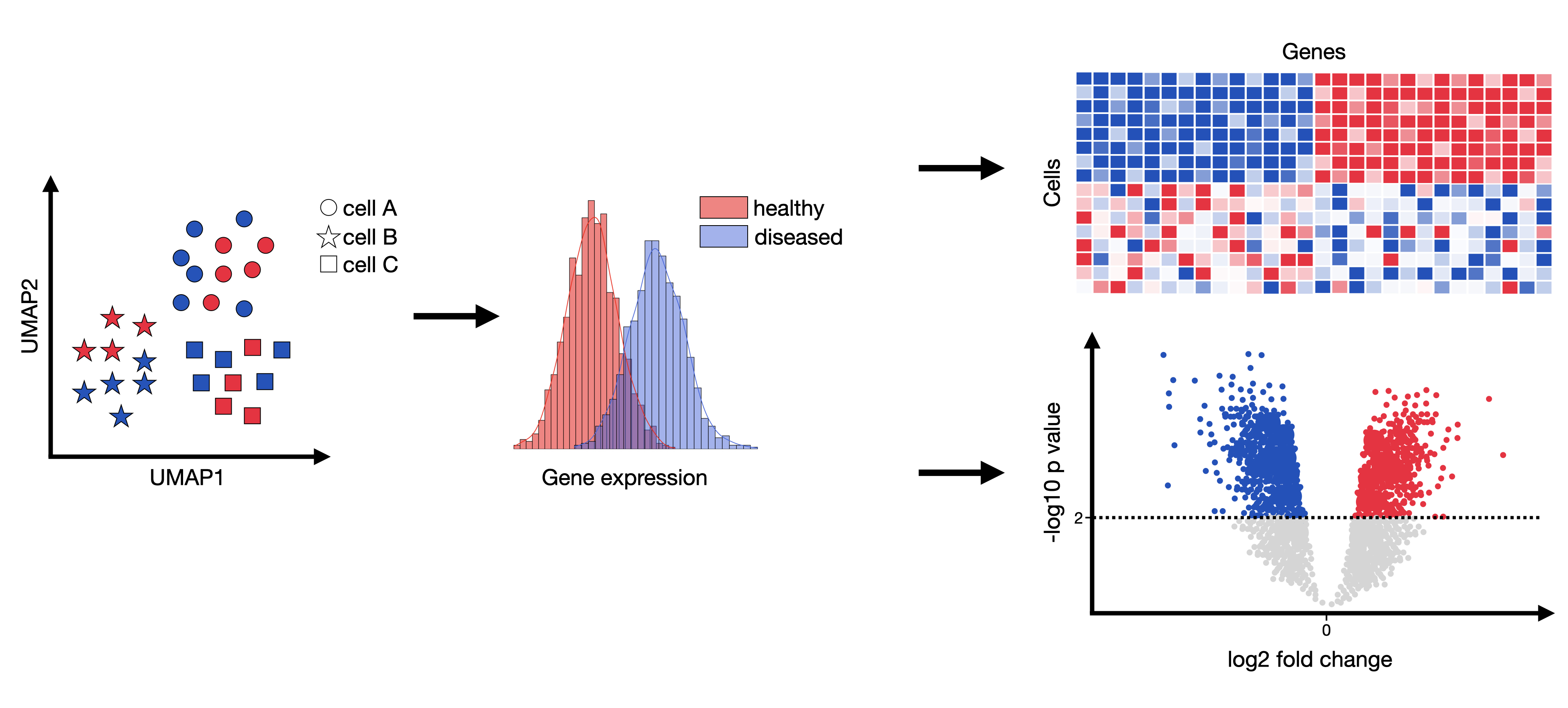
출처: link
개념
Differentially Expressed Genes (DEG) 분석은 두 개 이상의 샘플 그룹 간 유전자 발현 차이를 식별하는 과정이다. 이는 특정 조건이나 처리에 의해 유발된 유전자 발현 변화를 찾아내어 생물학적 메커니즘을 이해하는 데 도움을 준다.
예를 들어, 정상 세포와 암 세포 간의 유전자 발현 차이를 분석하여 암의 발병 메커니즘을 연구할 수 있다. DEG 분석을 통해 얻은 정보는 질병 진단, 치료 타겟 발굴 및 예후 예측 등에 활용될 수 있다.
가정
DEG 분석에서 중요한 전제 중 하나는 유전자가 다르게 발현하지 않는다는 귀무 가설(null hypothesis)이다.
이는 두 그룹 간 유의미한 차이가 없다고 가정하는 것이다.
분석의 목표는 이 귀무 가설을 기각하여, 유전자 발현에 유의미한 차이(Significant difference)가 있음을 증명하는 것이다.
이것은 DEG 분석뿐 아니라 두 집단 간 차이를 검정하는 모든 통계분석에서 쓰이는 가정이다.
이 귀무 가설을 기각하기 위해 유전자 발현에 대한 분포를 가정하고, 통계적 분석을 수행하게 된다.
물론, 비모수적 방법론 (non-parametric method)을 활용하여 분포를 따로 가정하지 않고, 두 그룹 간 유의한 차이를 보이는 유전자를 선별할 수도 있으나, 모수적 방법론들에 비해 많이 쓰이지 않는다.
- 모수적 방법론 기반 DEG 분석 도구
- EdgeR (2010): 36,217회
- DESeq2 (2014): 64,981회- 비모수적 방법론 기반
- NOISeq (2015): 736회
(google scholar 기반 피인용 횟수)
주요 가정 분포
RNA-seq 데이터의 특성상, 데이터가 정규 분포를 따르지 않으며 포아송(Poisson) 분포 또는 음이항(Negative Binomial) 분포를 가정하는 것이 일반적이다. RNA-seq 데이터는 카운트(Count) 데이터로서 각 유전자의 발현 수가 정수로 표현된다. 카운트 데이터는 자연스럽게 포아송 분포를 따르는 경향이 있지만, RNA-seq 데이터는 오버디스퍼션(Over-dispersion, 과도한 분산)을 나타내는 경우가 많아 음이항 분포를 사용하는 것이 더 적합하다.
"To account for biological variability, the negative binomial distribution has been used as a natural extension of the Poisson distribution, requiring an additional dispersion parameter to be estimated."
출처: From RNA-seq reads to differential expression results, Genome Biology, 2010 link
"For RNA-seq, it has been suggested that the Poisson distribution is well suited for analysis of technical replicates, whereas the higher variability between biological replicates necessitates a distribution incorporating overdispersion, such as the Negative Binomial."
출처: A comparison of methods for differential expression analysis of RNA-seq data, BMC Bioinformatics, 2013 link
"Negative binomial distribution has been shown to be a powerful model because it captures the over-dispersion of the nature of the count data generated by RNA-Seq experiments"
출처: Negative binomial additive model for RNA-Seq data analysis, BMC Bioinformatics, 2020 link
통계적 분석과 가정된 분포
그럼 위와 같이 가정된 분포를 어떻게 활용할까? 다시 귀무가설의 개념으로 돌아온다.
귀무가설과 대립가설
DEG 분석에서의 통계적 검정은 일반적으로 다음과 같은 귀무가설과 대립가설을 설정한다.
- 귀무가설(): 두 조건 간에 유전자 발현의 차이가 없다.
- 대립가설(): 두 조건 간에 유전자 발현의 차이가 있다.
이 가설 검정에서 각 그룹의 유전자 발현 데이터는 가정된 분포(예: 음이항 분포)를 따르는 것으로 모델링된다. 이를 통해 각 유전자에 대해 p-value를 계산하고, 유의미한 발현 차이를 보이는 유전자를 식별할 수 있다.
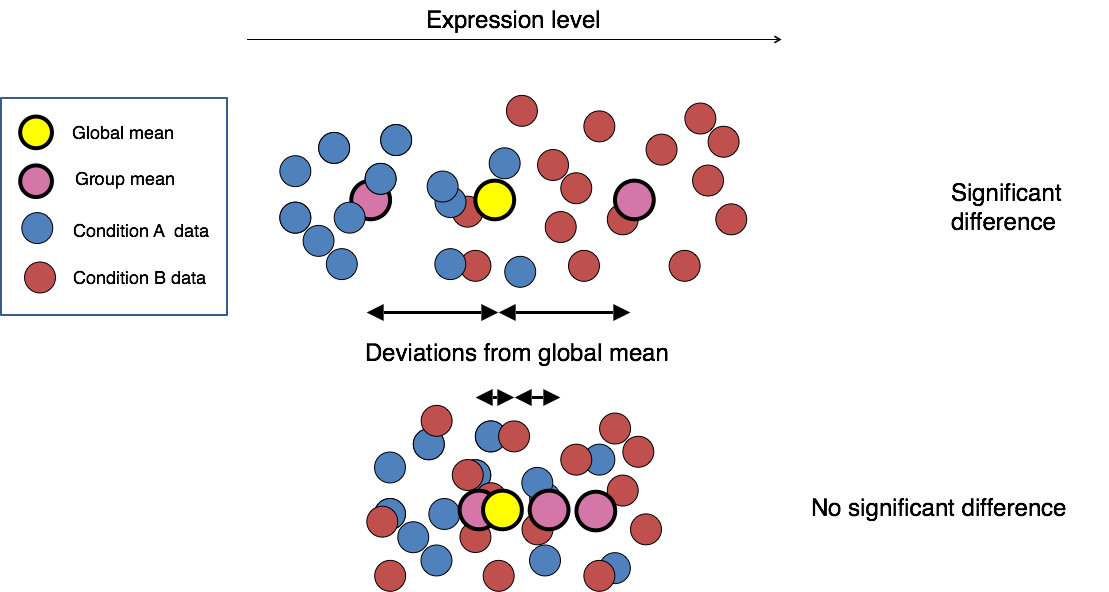
출처: link
가정된 분포를 기반으로 한 통계적 검정
조건이 서로 다른 그룹 A,B가 있다고 가정하고 예를 들어 보겠다.
1. 특정 유전자에 대한 그룹별 분포 모델링
- 그룹 A에 해당하는 샘플들의 유전자 'a' 발현값이 어떤 파라미터(성공확률 , 성공 횟수 )를 가진 음이항 분포를 따르는 지 추정.
- 마찬가지로, 그룹 B의 샘플들에서도 유전자 'a' 발현값이 어떤 파라미터를 따르는 음이항 분포를 따르는 지 추정. (즉, , 을 추정)
2. 두 분포 비교
- 두 그룹 간 유전자 'a'의 분포를 비교한다.
- 분포 비교는 결국 두 집단별로 추정된 파라미터 , 을 비교하는 과정이다.
- 일반적으로 음이항 분포에 대한 우도비 검정(Likelihood Ratio Test) 또는 왈드 검정(Wald Test)과 같은 방법을 사용하여 두 그룹의 파라미터 추정값 차이에 대한 통계적 유의성(p-value)을 계산한다.
- A의 , 과 B의 , 이 얼마나 다른지 p-value로 계산할 수 있다.
3. 유의미한 발현 차이 유전자 식별
- 이 과정을 전사체 전체에 대해 각 유전자별로 반복하여 p-value를 계산한다.
- 사전에 설정한 유의수준(예: 0.05) 이하의 p-value를 가지는 유전자를 유의미한 발현 차이를 보이는 유전자(DEG)로 식별한다.
- 다중검정 보정(FDR 등)을 적용하여 false positive rate를 제어할 수 있다.
요컨대, 가정된 분포(음이항 분포)를 바탕으로 각 유전자에 대해 두 그룹의 파라미터 추정값 차이에 대한 통계적 유의성을 계산하고, 이를 기반으로 발현 차이가 유의미한 유전자를 선별하는 과정이다.
Log2 Fold Change (Log2FC)
DEG 분석 결과를 해석할 때 p-value 외에도 log2FC (log2 fold change) 도 매우 중요한 지표로 활용된다. Fold change는 두 그룹 간 유전자 발현량의 상대적인 비율을 나타낸다.
Fold Change (FC) = (그룹 B의 평균 발현량) / (그룹 A의 평균 발현량)
Log2FC는 이 fold change 값에 밑이 2인 로그를 취한 값이다.
- Log2FC > 0 : 해당 유전자가 그룹 B에서 그룹 A보다 발현이 높음
- Log2FC < 0 : 해당 유전자가 그룹 A에서 그룹 B보다 발현이 높음
Log2FC의 절댓값이 클수록 두 그룹 간 발현 차이가 크다는 것을 의미한다. 일반적으로 |Log2FC| > 1 또는 2 정도를 기준으로 생물학적으로 의미 있는 발현 차이라고 간주한다.
p-value와 Log2FC를 함께 고려하여 DEG를 선별하는 것이 일반적이다. 예를 들어 p-value < 0.05 이면서 |Log2FC| > 1 또는 2인 유전자를 DEG로 지정하는 식이다.
p-value와 Log2FC의 역할
p-value는 통계적 유의성을 나타내는 지표인 반면, fold change(FC)는 두 그룹 간 실제 발현량 차이의 정도를 보여주는 지표이다.
p-value는 두 그룹 간 발현량 차이가 우연에 의한 것인지를 판단하는 데 사용되며, 작을수록 그 차이가 우연이 아닌 유의미한 차이일 가능성이 높다. 반면 fold change는 그 발현량 차이의 생물학적 크기를 나타낸다. 큰 fold change 값은 두 그룹 간 실제 발현량 차이가 크다는 것을 의미한다.
따라서 DEG 분석에서는 p-value와 fold change 두 가지 기준을 모두 고려하는 것이 일반적이다. 예를 들어 p-value < 0.05이면서 |log2FC| > 1 또는 2 같은 조건을 만족하는 유전자를 DEG로 선별한다.
- p-value는 통계적 유의성을 판단하는 기준
- Fold change는 실제 발현량 차이의 생물학적 크기를 나타내는 기준
이렇게 p-value와 fold change의 역할을 구분하여, 두 가지를 모두 활용해야 의미 있는 DEG 분석 결과를 얻을 수 있다.
통계적 가정을 위한 데이터 전처리 필요성
앞서 설명한 바와 같이, DEG 분석에서는 '두 그룹 간 유전자 발현에 차이가 없다' 는 귀무가설을 세운 뒤, 이를 기각할 수 있는지 통계적으로 검정한다.
이 과정에서 유전자 발현 데이터가 특정 분포를 따른다고 가정한다(예: 음이항 분포).
그런데 실제 RNA-seq 데이터에는 다양한 기술적, 생물학적 요인들이 영향을 미친다.
"Raw read counts alone are not sufficient to compare expression levels among samples, as these values are affected by factors such as transcript length (유전자 길이), total number of reads (라이브러리 크기), and sequencing biases."
(출처: link / 정규화 관련 참고자료: link)
이를 적절히 보정하지 않으면 분석 결과의 정확성이 크게 떨어질 수 있다. 따라서 DEG 분석에 앞서 몇 가지 분석이 추가로 필요하다.
1. 데이터 품질 관리 (QC)
RNA-seq 데이터에는 다양한 기술적 요인(technical bias)들이 포함되어 있다. 예를 들어 서열 읽기 오류, 어댑터 서열 오염, GC 함량에 따른 편향 등이 있다. 만약 이러한 기술적 요인들을 제거하지 않고 DEG 분석을 하면 결과의 정확성이 크게 떨어질 수 있다. 따라서 트리밍, 필터링 등의 전처리 단계를 거쳐 기술적 요인을 최소화하고 데이터의 품질과 신뢰성을 높여야 한다. 전처리를 통해 기술적 노이즈를 제거하고 (내가 탐색하고 싶은) 실제 생물학적 신호를 강화할 수 있다.
2. 라이브러리 크기 조정 (Normalization)
샘플 간 총 읽기(read) 수 (라이브러리 크기, library size)의 차이를 정규화(normalization)하지 않으면, 귀무가설 검정과 log2FC 값 해석에 오류가 발생한다. 라이브러리 크기 차이에 의한 발현량 변화까지 유전자 발현 차이로 간주하게 되어, 귀무가설을 잘못 기각하거나 log2FC 값이 왜곡될 수 있다.
3. 유전자 길이 보정
긴 유전자(정확히는 transcript)일수록 더 많은 읽기(read)가 매핑되므로, 단순히 읽기 수를 사용하면 유전자 간 발현량 차이를 제대로 비교하기 어렵다. 예를 들어, 특정 유전자보다 길이가 2배 긴 유전자라면 동일한 발현량에도 긴 유전자가 2배 많은 읽기 수를 보일 것이다. 따라서 유전자 길이에 따른 이런 편향을 보정하지 않으면, 유전자 간 발현 차이 비교가 왜곡될 수 있다. 이를 위해 일반적으로 RPKM, FPKM, TPM 등의 정규화 지표를 사용하여 유전자 길이로 인한 영향을 제거한다. 단, 분석 목적과 조건에 따라 유전자 길이 보정의 필요성이 달라질 수 있다. 예를 들어 동일 유전자에 대한 그룹 간 직접 비교인 경우에는 길이 보정이 크게 중요하지 않을 수 있다.
이처럼 DEG 분석 시 이러한 전제와 가정들을 고려해야 한다. 이를 모두 잘 고려해야만 통계적 가설 검정 결과(p-value)와 실제 발현 차이의 크기(log2FC)를 신뢰할 수 있다. DEG 분석의 정확성을 높이기 위해서는 데이터의 특성을 제대로 반영한 전처리와 정규화가 선행되어야 한다.
추가로.. single cell RNA-sequencing 에서는 음이항 분포뿐 아니라, 영과잉 음이항 (Zero-inflated Negative bionomial, ZINB) 분포를 가정하는 경우도 있다.
(Sparsity 문제를 해결하기 위함이다. bulk RNA-seq에 비해 scRNA-seq은 '0'인 유전자가 상당히 많다.)
두 분포를 가정하였을 때, DEG 결과에서 어떤 차이가 있는 지에 대해 탐구한 연구도 있는데, 이는 나중에 다루겠다.
To do: 모수적vs.비모수적 통계방법론 / (Zero-inflated) 음이항 분포와 포아송 분포 / single cell RNA-seq DEG 분석 방법론 / RNA-seq 전처리 / RNA-seq 발현량 정규화 / 간단한 DEG 파이프라인 및 예제 코드

질문이 몇 가지 있습니다.
1. 두 분포에 대한 유전자 발현량이 어떤 파라미터(p, r)를 가진 음이항 분포를 따르는지 추정하는 것이라고 하셨는데, 어떤 파라미터 p, r의 성공확률, 성공횟수에서 '성공'이라는 사건은 어떤 사건을 의미하는 건가요..?
2. DESeq2가 edgeR에 비해 2배 더 많이 사용되는데 특별한 이유가 있을까요? 두 가지 tool 중에 DESeq2가 조금 더 선호되는 특별한 이유가 있는지 궁금합니다.