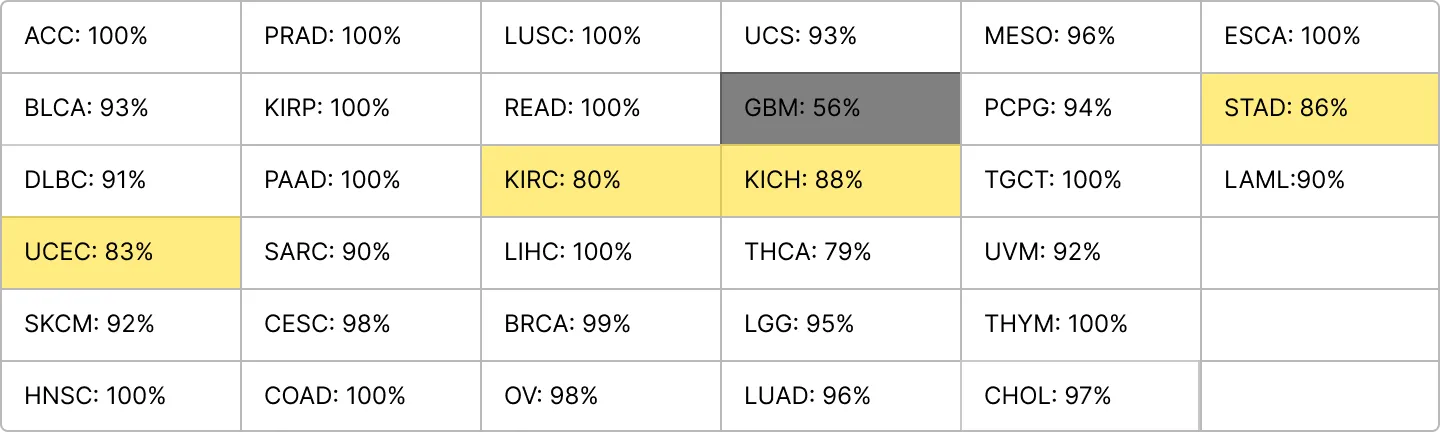
결과가 기묘하게 나왔다.
Clustering이랑 GO_slim방식에서 Clustering이 약 1~2% 성능이 더 좋았다.
원래는 GO_slim의 해석 가능성을 어떻게든 다른곳에서 활용하고 싶었는데 성능이 Clustering방식이 더 좋게 나왔다. 막말로 해석 가능성의 기능을 쓰지 못한다는 것이다.
Clustering
ACC = 94%
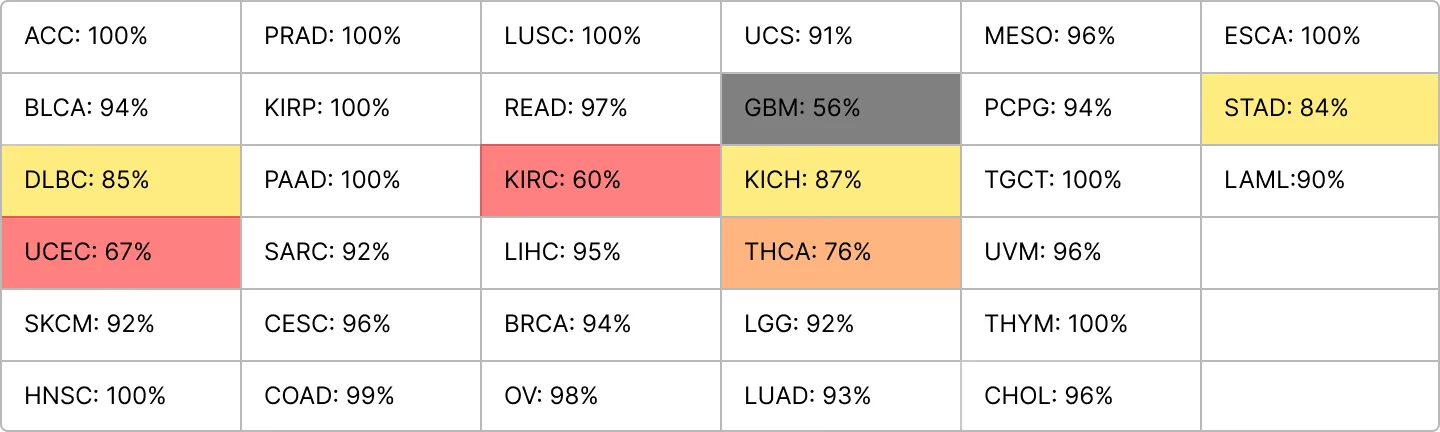
GO_Slim
ACC = 92.6%
왤까...? 라는 의문을 가지다가 go의 경우에는 go term이 있는 json에 해당 유전자가 없거나 go term내의 유전자 갯수가 특정 갯수를 넘지 않으면 이를 버리고 사용해서 버리는 것들 중에 중요한 유전자가 있다는 생각이 문득 들었다.
def gene_ontology_slim_grouping(
in_features: int,
proj_size: int,
n_group: int,
train_data: DataFrame,
drop_remainder: bool = True,
strategy: str = "split",
min_size: int = 200,
max_size: int = 500,
**kwargs,
) -> Tuple:
# min_size = kwargs.get("min_size", min_size)
assert (
max_size > min_size
), "[GO Slim] `max_size` cannot be lower or equal than `min_size`"
logger.info(
"When grouping features with GO Slim, `n_group` or `group_size` parameters"
+ " are ignored. The number of group will depend on the `strategy` and the `group_size`"
+ f" will be `min_size`={min_size}"
)
name_to_pos = {
name.split(".")[0]: pos for pos, name in enumerate(train_data.columns)
}
num_columns = train_data.shape[1]
file_path = Path("go_bp_gene_graph.json")
with file_path.open("r") as f:
slim_groups = json.load(f)
if not drop_remainder:
logger.warning("Using GO slims with unmapped genes is not yet supported")
pass
group_name = []
idx_in = []
n_groups = 0
non_valid_go_term_no = 0
valid_go_term_no = 0
used_genes = set()
total_genes = set()
if strategy == "split":
for go_term, genes in slim_groups.items():
n_genes = len(genes["genes"])
valid_genes = []
missing_genes = []
for gene in genes["genes"]:
pos = name_to_pos.get(gene)
total_genes.add(gene)
if pos is not None:
valid_genes.append(pos)
used_genes.add(gene)
else:
missing_genes.append(gene)
genes_tensor = torch.tensor(valid_genes)
n_valid_genes = len(valid_genes)
# print(f"valid_genes_nums: {n_valid_genes}")
if n_valid_genes >= min_size:
valid_go_term_no += 1
# print(f"{go_term} 유효 유전자 {n_valid_genes}개 / 비율: {round((n_valid_genes/n_genes),2) * 100}% ")
if n_valid_genes >= max_size * 1.5: # because of rounding strategy
split_groups = round(n_valid_genes / max_size)
# print(f"{go_term} has surpassed maximum {max_size} size, total {n_genes} of valid {n_valid_genes} genes has split to {split_groups} groups.")
# print("=============================")
group_name.extend(
[f"{go_term}-{i}" for i in range(1, split_groups + 1)]
)
chunk_sizes = (n_valid_genes // split_groups) + (
np.arange(split_groups) < (n_valid_genes % split_groups)
)
genes_split = genes_tensor.split(chunk_sizes.tolist(), dim=0)
idx_in.extend(genes_split)
n_groups += split_groups
else:
group_name.append(go_term)
idx_in.append(genes_tensor)
n_groups += 1
else:
# print(f"{go_term} is not valid term")
non_valid_go_term_no += 1
grp_proj_dim = [[min_size] for _ in range(n_groups)]
print("==========================")
print(f"JSON 전체 유전자: {len(total_genes)}개")
print(f"사용된 유전자 갯수: {len(used_genes)}개")
# print(f"JSON에서 사용된 유전자 비율: {round((len(used_genes)/len(total_genes)),2)*100}%")
# print(f"TCGA 데이터에서 사용된 유전자 비율: {round((len(used_genes)/num_columns),2)*100}%")
print(f"사이즈 조정: {min_size}")
print(f"유효 GO term 갯수: {valid_go_term_no}")
# print(f"그룹 이름: {group_name}")
print(f"유효하지 않은 GO term 갯수: {non_valid_go_term_no}개" )
print("==========================")이미 수정된 코드지만 저기서 사용되는 min_size를 통해 들어갈 go- term을 정하게 되는 거라 connectivity_kwargs를 통해 모델을 설정할때 여러가지 사이즈로 조정해서 모델을 돌려보았다.
model = AttOmics(
n_group= 10,
n_layers= 1,
num_heads= 1,
attention_norm= "layer_norm",
grouping_method= "gene_ontology_slim",
head_norm= "layer_norm",
sa_residual_connection= True,
head_residual_connection= False,
head_dropout= 0.0,
head_batch_norm= False,
reuse_grp= True,
constant_group_size= False,
head_input_dim= 500,
head_hidden_ratio=[0.5],
input_dim=X.shape[1], # a dict of dimension
num_classes=n_class,
label_type="cancer_type",
class_weights=class_weights,
train_data=X,
optimizer_init=optimizer,
scheduler_init=lr_scheduler,
connectivity_kwargs={"min_size": 100},
generate_ATM = False,
generate_CFM = False,
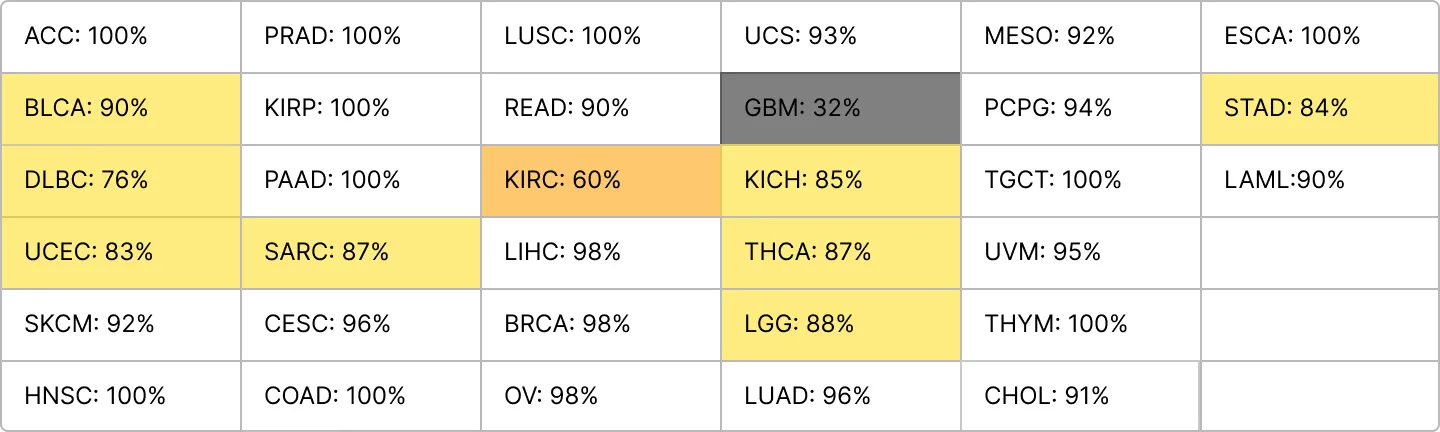
epoch_graph = True)min_size가 작을수록 더 많은 유전자가 들어갈거라 생각해서 성능이 더 좋아질거라 생각했는데.....
min_size = 150
ACC = 82%
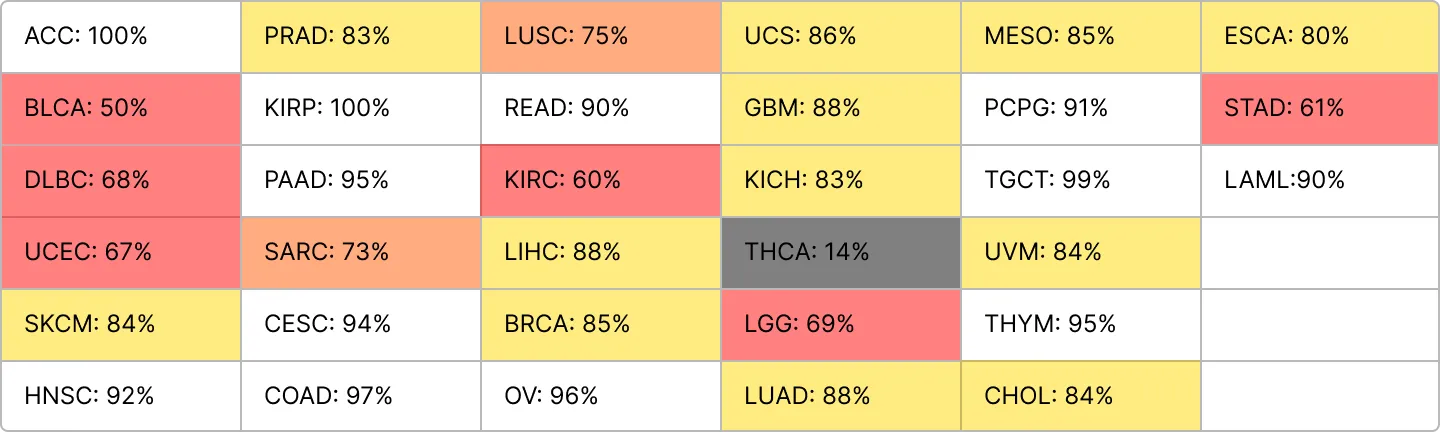
min_size = 100
ACC = 88%
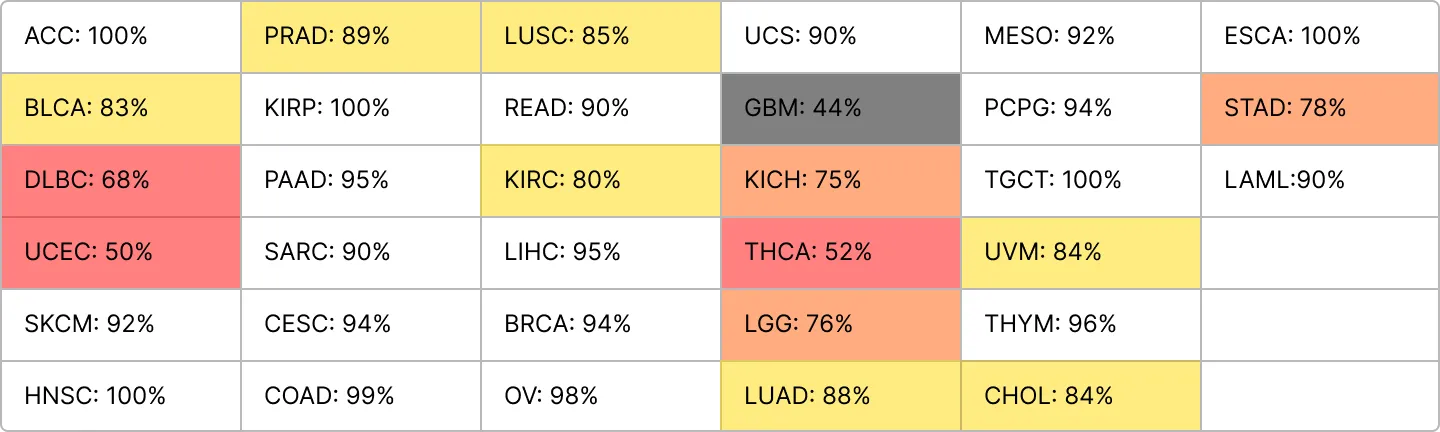
min_size = 50
ACC = 88%
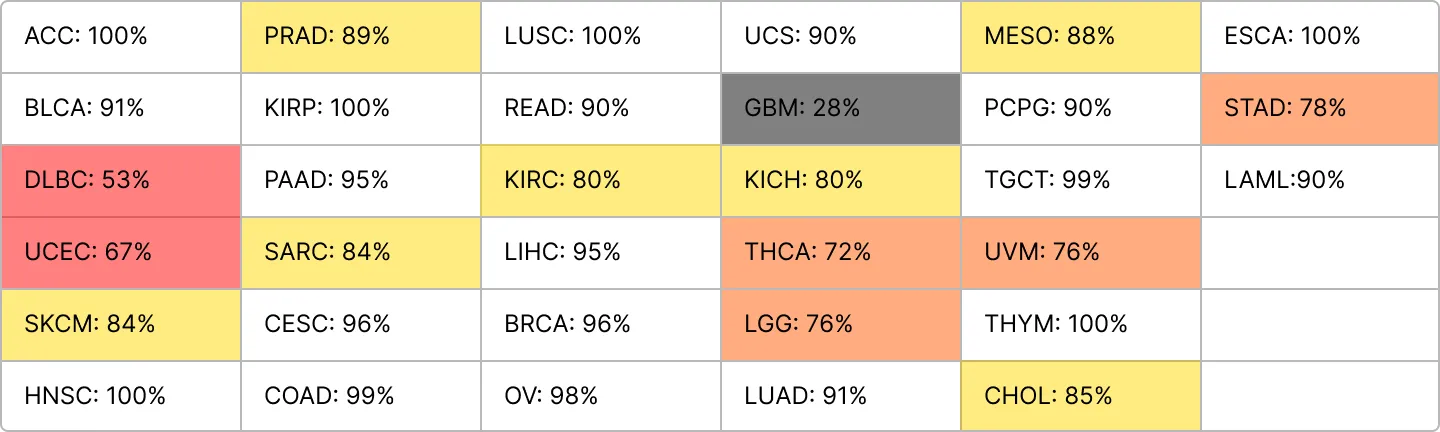
min_size가 기본으로 200으로 정해져 있는데 왜 150으로 줄때 다시 정확도가 줄어드는걸까....?
진짜 모르겠다.

인간입니다. 다만 컴공을 전공하고있는...