결국 전 포스트에서 오류가 떳던 이유는 단순했다.
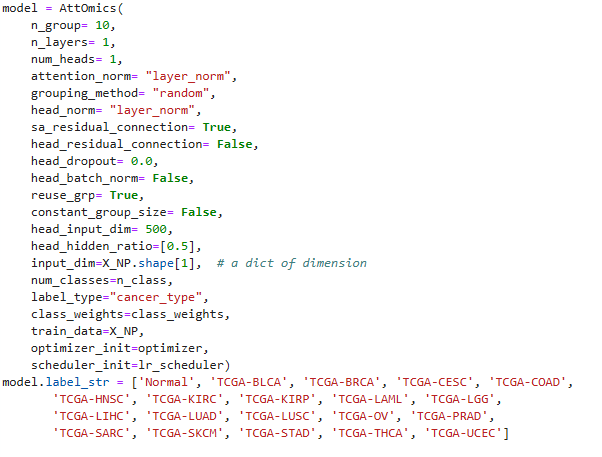
data loader 코드와 위의 사진의 model 선언에 있는 train_data부분이 같지 않아서 계속 오류가 떳었다.
이후에 주어진 데이터로는 GO를 사용하는 방법을 못쓰기 때문에 일단 교수님이 주신 TCGA-BRCA 데이터에 대해 돌려보았다.
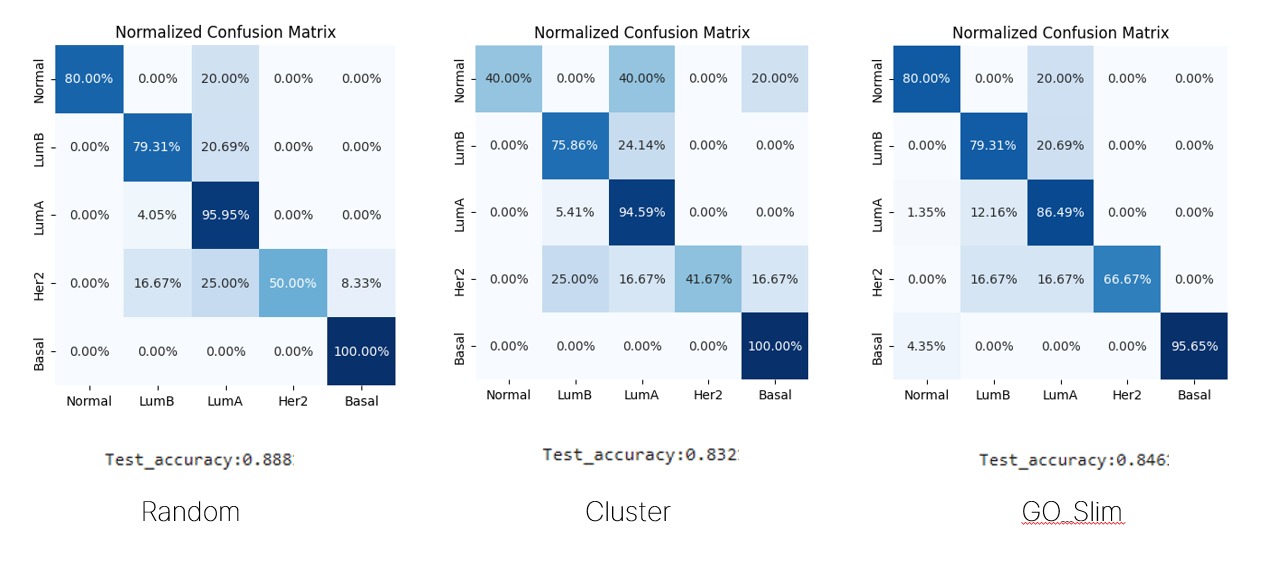
Her2에 대한 classification이 너무 안되길래 일단 최근에 나온 논문들중에 분류가 잘되는 것들이 있는지 찾아보다가
Multi-Omic Graph Diagnosis (MOGDx)라는 이 논문을 찾았다. 물론 single omics에 대한 모델은 아니지만 여러가지 omics 데이터들을 좀 합치면 좋은 성능의 질병 classification이 가능하고 + 해석에 대한 가능성을 주긴 했지만...
솔직히 해석에 대한 것들은 너무 끼워맞추기 느낌이 강했다. 내가 뭘 알겠냐 하겠지만 해석의 방식이 좀 의아하다....
물론 여러가지 omics 데이터들을 합쳐서 사용할 수 있다는게 좀 의미가 크다 생각하지만 해석의 면에서는 오히려 기존에 진행하고 있던 AttOmics가 단일 종류의 데이터에서 weight를 확인해볼수 있기 때문에 일단 이걸로 마저 진행하기로 하고 데이터만 LGG, KIPAN 으로 비교 대상군을 늘리기로했다.
근데 해당 논문에서 R 스크립트로 다운받게 하는데 R서버가 잠시 문을 닫았는지 안들어가진다....
어제 새벽에 서버가 점검을 하고있었는지 접속이 안되서 library 다운을 하지 못했다.
심지어 접속 이후에 새벽에 다운을 하는데 xfun, knitr, rmarkdown 패키지들이 아래와 같은 오류들이 반복적으로 계속 발생해서 뭘하지도 못했다.
Warning in install.packages : installation of package ‘rmarkdown’ had non-zero exit status
namespace ‘xfun’ 0.43 is being loaded, but >= 0.44 is required
관련 오류 사항들도 링크들을 보면 해당 package들을 그냥 삭제 했다가 다시 다운 받고 그러라 했지만 dependency들이 꼬였는지 나한테는 작동하지 않았고 수동으로 tar.gz파일을 받아 rtools로도 해봤었지만 나한텐 해당사항이 아니였었다.
결국 최후의 방법으로 논문에서 요구하는 4.2.3 버전의 R을 싹다 지워버리고 그냥 가장 최신 버전인 4.4.1 버전을 사용해서 다시 다 다운 받았다.
install.packages("reshape2")
install.packages("kableExtra")
install.packages("plotly")
install.packages("pheatmap")
if (!require("BiocManager", quietly = TRUE))
install.packages("BiocManager")
BiocManager::install("vsn")
BiocManager::install("SummarizedExperiment")
library(reshape2)
library(kableExtra)
library(plotly)
library(vsn)
library(tibble)
library(pheatmap)
library(SummarizedExperiment)
print("Preprocessing lib DONE")
install.packages('remotes')
remotes::install_github('marioni-group/methylpiper')
install.packages("igraph")
BiocManager::install("GO.db")
BiocManager::install("impute")
BiocManager::install("DESeq2")
BiocManager::install("edgeR")
install.packages("WGCNA")
library(MethylPipeR)
library(glmnet)
library(igraph)
library(DESeq2)
library(dplyr)
library(ggplot2)
library(edgeR)
library(MethylPipeR)
library(WGCNA)
print("preprocess_functions lib Done")
단순히 CRAN package 뿐만 아니라 BiocManager등 별의별 package들을 끌어와서 사용하는 거였기에 구글링하면서 필요한 것들을 적용시켜서 다운받게 했더니 필요한 패키지들을 모두 적용 시킬 수 있었다.
각종 패키지들은 아래의 링크로 남겨놓았다. 종종 CRAN 패키지를 다운받을려면 필요 패키지들도 요구하는경우가 있기 때문에 한번 확인해볼것
한 예시로 CRAN의 WCGNA 패키지를 다운받는데 필요한 것들은 아래 사진에 import에 있는 패키지들을 요구한다.

나의 경우에는 GO.db 가 없어서 해당 패키지 단어를 눌러보니 아래처럼 나왔다.
그래도 R 터미널에서 해당 installation 코드를 실행하면 되는 식.
