이전 포스트에서 말한 TCGA데이터는 크게 3개 였다 BRCA, KIPAN, LGG.
BRCA랑 LGG의 경우 단일 데이터라서 그냥 다운받은 대로 깃헙에 있는대로 해주면되는데 KIPAN의 경우 KICH, KIRP, KIRC 3개의 데이터를 합쳐서 하나의 데이터로 만들어야 한다.
너무 간만에 R을 만지는 거라 다시 익숙해지는데 좀 걸렸고, 이에 맞는 환경도 만드느라 애를 먹었다.
KIPAN_CONCAT.r
# 프로젝트와 데이터셋 정의
project <- 'KIPAN'
dataset <- 'TCGA'
modal1 <- 'KIRC'
modal2 <- 'KIRP'
modal3 <- 'KICH'
trait <- 'subtype'
source('./preprocess_functions.R')
datMeta1 <- read.csv(paste0('./data/TCGA-',modal1,'/datMeta.csv') , row.names = 1)
datMeta2 <- read.csv(paste0('./data/TCGA-',modal2,'/datMeta.csv') , row.names = 1)
datMeta3 <- read.csv(paste0('./data/TCGA-',modal3,'/datMeta.csv') , row.names = 1)
# 첫 번째 데이터셋 처리
KIRC <- load(paste0('./data/TCGA-', modal1, '/mRNA/mRNA.rda'))
count_mtx1 <- assay(get(KIRC))
colnames(count_mtx1) <- substr(colnames(count_mtx1), 1, 12)
count_mtx1 <- count_mtx1[, !(duplicated(colnames(count_mtx1)))]
common_idx1 <- intersect(colnames(count_mtx1), rownames(datMeta1))
count_mtx1 <- count_mtx1[, common_idx1]
datMeta1 <- datMeta1[common_idx1, ]
#=======================================================================
# 두 번째 데이터셋 처리
KIRP <- load(paste0('./data/TCGA-', modal2, '/mRNA/mRNA.rda'))
count_mtx2 <- assay(get(KIRP))
colnames(count_mtx2) <- substr(colnames(count_mtx2), 1, 12)
count_mtx2 <- count_mtx2[, !(duplicated(colnames(count_mtx2)))]
common_idx2 <- intersect(colnames(count_mtx2), rownames(datMeta2))
count_mtx2 <- count_mtx2[, common_idx2]
datMeta2 <- datMeta2[common_idx2, ]
#==============================================================
#3번째 데이터셋
KICH <- load(paste0('./data/TCGA-', modal3, '/mRNA/mRNA.rda'))
count_mtx3 <- assay(get(KICH))
colnames(count_mtx3) <- substr(colnames(count_mtx3), 1, 12)
count_mtx3 <- count_mtx3[, !(duplicated(colnames(count_mtx3)))]
common_idx3 <- intersect(colnames(count_mtx3), rownames(datMeta3))
count_mtx3 <- count_mtx3[, common_idx3]
datMeta3 <- datMeta3[common_idx3, ]
#combine===================================================
count_mtx <- cbind(count_mtx1, count_mtx2, count_mtx3)
datMeta <- rbind(datMeta1,datMeta2,datMeta3)
diff_expr_res <- diff_expr(count_mtx , datMeta , trait , 500 , 'mRNA')
# Save differential expression results
datExpr <- diff_expr_res$datExpr
datMeta <- diff_expr_res$datMeta
dds <- diff_expr_res$dds
top_genes <- diff_expr_res$top_genes
save(datExpr, datMeta, dds, top_genes, file=paste0('./data/',dataset,'/',project,'/mRNA_processed.RData'))
그리고 후에 저장된 CSV에는 subtype들이 따로 없었기에 환자에 맞는subtype를 추가해 새로 저장, 그리고 유전자 이름들이 아닌 유전자 char로 되어있었기 때문에 이것까지 모두 적용해서 최종 csv를 만들어주었다.
add_subtype.r
# Load the required data
datExpr <- read.csv(paste0('./data/TCGA/BRCA/raw/datExpr_mRNA.csv'), row.names = 1)
datMeta <- read.csv(paste0('./data/TCGA/BRCA/raw/datMeta_mRNA.csv'), row.names = 1)
# Ensure datExpr columns match patient IDs in datMeta
patient_ids <- colnames(datExpr)
patient_ids <- gsub("\\.", "-", patient_ids)
if (!all(patient_ids %in% rownames(datMeta))) {
stop("Patient IDs in datExpr do not match those in datMeta.")
}
# Extract subtype information from datMeta
subtype_vector <- datMeta[patient_ids, "paper_BRCA_Subtype_PAM50"]
# Add SUBTYPE row to datExpr
datExpr["paper_BRCA_Subtype_PAM50", ] <- subtype_vector
datExpr_transposed <- t(datExpr)
# Convert to data frame for easier manipulation
datExpr_transposed_df <- as.data.frame(datExpr_transposed)
# Save the updated datExpr with SUBTYPE
write.csv(datExpr_transposed_df, file = ('./data/TCGA/BRCA/raw/datExpr_mRNA_with_pam.csv'), row.names = TRUE)
print("datExpr with SUBTYPE has been saved.")
참고로 유전자 char <> 유전자 이름을 교환하기 위해서는 관련 테이블이 있어야하는데 r에서 해당 tcga 데이터의 rda파일을 불러와 rowData(data)를 하면 나오기때문에 필요한 부분만 때와서 새로 csv에 저장을 해주었다.
gene_mapping.py
import pandas as pd
tcga_name = 'LGG'
gene_table = pd.read_csv('./data/TCGA/'+ tcga_name + '/'+ tcga_name + '_GENE_NAME.csv')
dat = pd.read_csv('./data/TCGA/'+ tcga_name + '/raw/datExpr_mRNA_with_grade.csv')
gene_mapping = dict(zip(gene_table['gene_id'], gene_table['gene_name']))
# print(gene_mapping)
new_columns = [gene_mapping.get(col, col) for col in dat.columns]
print(new_columns)
print(len(new_columns))
dat.columns = new_columns
dat.to_csv('./data/TCGA/'+ tcga_name+ '/TCGA_'+tcga_name+'.csv', index=False)
그렇게 3개의 TCGA-BRCA/KIPAN/LGG 3개의 파일을 만들 수 있었다.
각각의 subtype들은 아래와 같다.
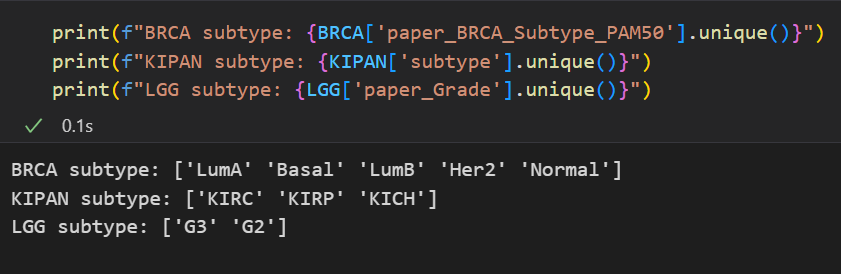
이전에 가지고 있던 BRCA에 대해서 교수님이랑 상의를 해봤지만 MOGDx에서 Pre-procssing까지 하는 데이터의 경우에도 거의 raw 데이터인거 같다고 하셔서 그냥 이걸로 사용해도 된다 하셨다.

인간입니다. 다만 컴공을 전공하고있는...