잠시 하라는 코드 수정및 결과 산출은 안하고 putty 커스터마이징하느라 시간을 사용했다.

사실 기본 putty 설정이 좀 눈깔이 빠져나갈거같았다. 글씨 크기도 그렇고 글꼴도 그렇고 가독성이 최악이고 안그래도 안경쓰고 있는데 거북목이 더 심해지고 싶지 않아져서 문득 시간을 할당해서 좀 바꾸었다.
다음부턴 절대 Courier new 글꼴은 죽어도 쓰지 않는걸로...
putty 에 colours >> putty displays이랑 ./bashrc를 통해 적당히 프롬포트 색 변경으로 적당히 설정해주었다.
각설하고 코드를 돌리려고 했는데 자꾸 아래와 같은 오류가 떳었다.
RuntimeError: split_with_sizes expects split_sizes have only non-negative entries, but got split_sizes=[455, -28]
내가 끄적거린 코드를 찾다가 문제점을 찾아냈다.
아래의 코드 부분에서 추가 코드라는 주석부분에 코드가 이상했었다.
애초에 만들었을때 전체 유전자 중에서 valid_genes랑 missing_genes를 GO BP를 통해서 걸러냈는데 그냥 전체 유전자를 넣어버려서 갯수가 음수가 나버렸다.
확인해보니 전체유전자는 455개 , valid_genes는 427개가 나와서 빼고 나서 -28이 나오게 된것.
if n_genes >= min_size:
if n_genes >= max_size * 1.5: # because of rounding strategy
split_groups = round(n_genes / max_size)
logger.info(
f"Go term {go_term} has been split in {split_groups} groups."
)
group_name.extend(
[f"{go_term}-{i}" for i in range(1, split_groups + 1)]
)
chunk_sizes = (n_genes // split_groups) + (
np.arange(split_groups) < (n_genes % split_groups)
)
# print("=============================")
# print(f"청크사이즈: {chunk_sizes}")
# print(f"텐서사이즈: {genes_tensor}")
#추가코드
if sum(chunk_sizes) != len(genes_tensor):
print("wtf")
chunk_sizes[-1] += len(genes_tensor) - sum(chunk_sizes)
genes_split = genes_tensor.split(chunk_sizes.tolist(), dim=0)
idx_in.extend(genes_split)
n_groups += split_groups
else:
group_name.append(go_term)
idx_in.append(genes_tensor)
n_groups += 1진짜 어이없는 실수를 해서 그냥 n_genes대신에 valid_genes 갯수를 변수 선언해서 대체해주고 해당 추가코드는 없앴다.
TCGA_BRCA의 경우 잘 돌아갔고 TCGA_KIPAN을 돌리려했는데 또하나의 오류가 떳었다.
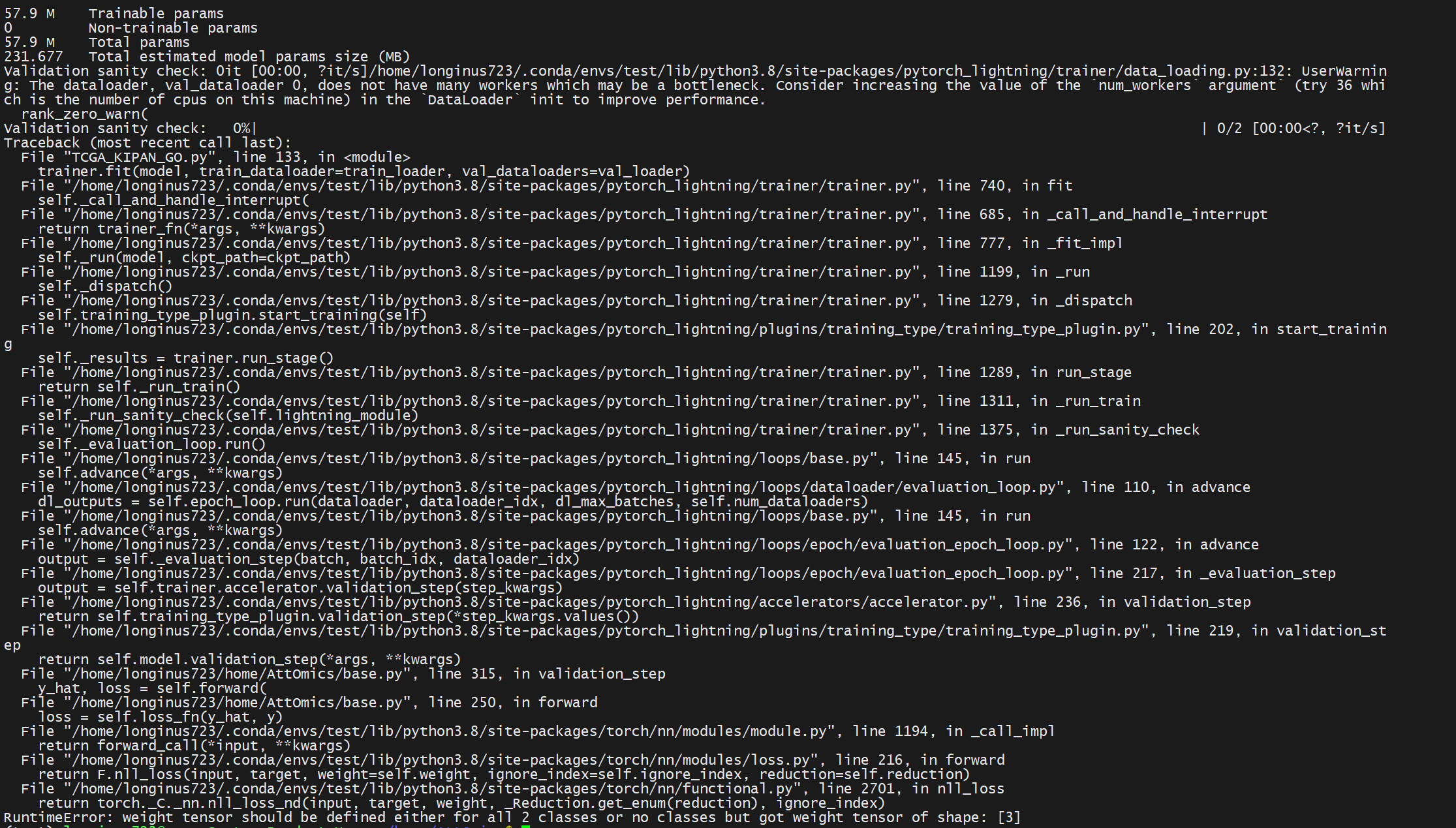
원래 Classification 3개 였는데 어째서인지 dataload를 직접 돌릴때는 2개의 class밖에 없다는 것이다. 다시 csv 불러온거 확인해보고 KIPAN의 subtype까지 제대로 있었는데 자꾸 2개 밖에 없다는 오류가 뜨길래 뭐지 싶었다.
KIPAN의 경우 R 파일에서 내가 만들때 KICH, KIRP, KIRC를 합치는 과정에서 그냥 쭉 이어붙이는 방식으로 CSV를 저장을했었다는게 문득 생각났다.

그리고 확인해보니 train/valid/test를 나눌때 그냥 위에서부터 순서대로 나눈거라 '아 전체의 70% 정도면 마지막 subtype이 포함 안될 법하구나' 라는 생각이 문득 들었고, 확인해보니 KICH이 label이 train에서는 없었다는게 확인되었다.
그래서 그냥 단순하게 귀찮아서 행을 기준으로 무작위로 섞은 DataFrame을 만들어서 적용시켜주었다.

혹시몰라 무작위로 행을 섞지 않았던 TCGA_BRCA, TCGA_LGG에서도 영향을 끼쳤을까봐 똑같이 적용을 해주었다. 실제로 둘다 성능이 향상되었다.
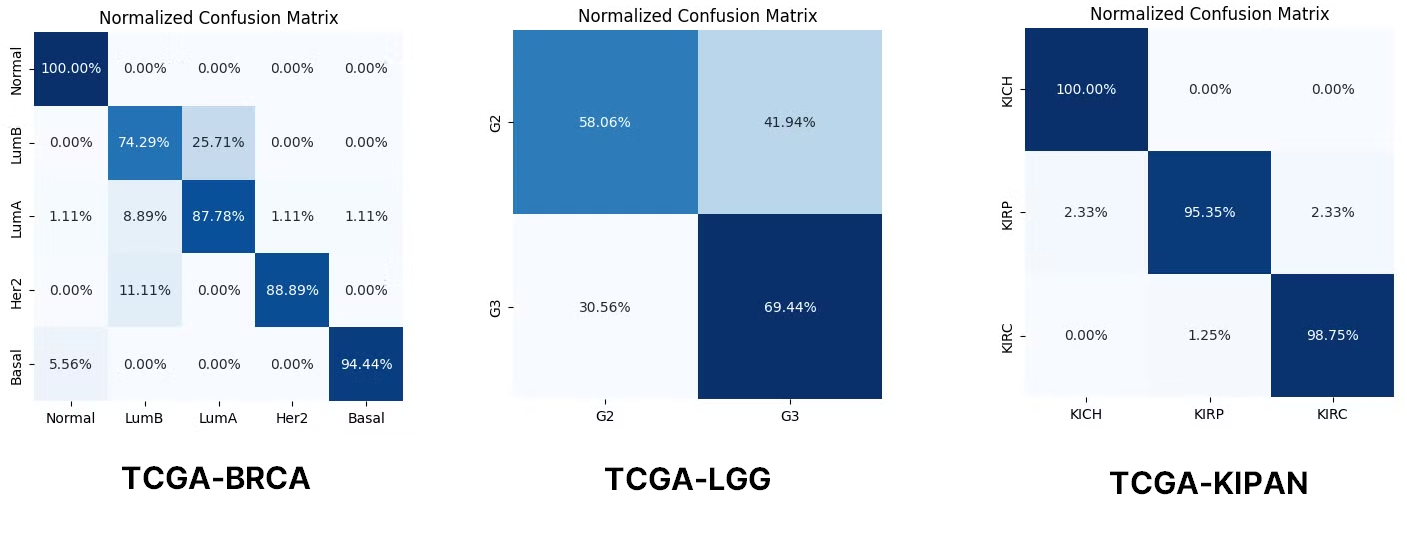
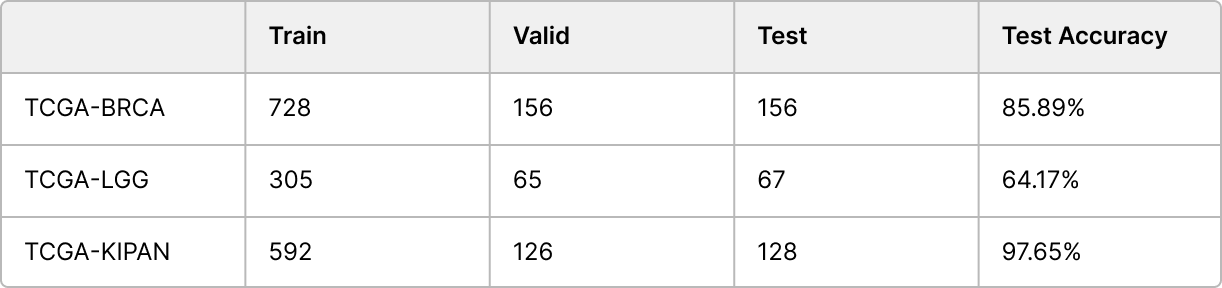
BRCA랑 KIPAN의 경우는 성능이 괜찮게 나왔지만 LGG는 정확도가 좀 낮은거같다. 아마도 LGG의 경우는 Train 갯수가 아래의 표처럼 너무 적어서 Under-fitting으로 인해 정확도가 좀 낮게 나온게 아닐까 생각된다.