들어가기에 앞서 SMILE 화학식 코드에 대해서 유용한 라이브러리를 알개되었다
RDKit
RDKit이라는 라이브러리로, SMILES ↔ 분자 객체(Mol) 변환이 가능하며, 다양한 분자 지문(fingerprint), 분자 지표(descriptor) 계산을 할 수 있다.
from rdkit import Chem
mol = Chem.MolFromSmiles('CCO') # SMILES → 분자 객체
smi = Chem.MolToSmiles(mol) # 분자 객체 → SMILES
fp = Chem.RDKFingerprint(mol) # 분자 지문 생성smiles_list = [
'CCOc1ccc(CC(=O)O)cc1', # 아세트아닐리드 유사
'CCN(CC)CCOC(=O)c1ccccc1', # 베타 차단제 유사
'C1=CC=C2C(=C1)C=CC=C2', # 나프탈렌 (비약물)
]
for mol, smi in zip(mols, smiles_list):
if mol is None:
continue
mw = Descriptors.ExactMolWt(mol) # 정확한 분자량
logp = Crippen.MolLogP(mol) # clogP
qed_s = QED.qed(mol) # QED score (0~1)
results.append((smi, mw, logp, qed_s))위의 코드와 같이 스마일 코드를 입력하면, 분자량과 Fingerprint 정보 등을 알 수 있는 유용한 라이브러리이다.
데이터 분석을 진행하는 과정에서, 내가 진행했던 코드가 사라져 다른 팀원분들의 코드를 참고하였다.
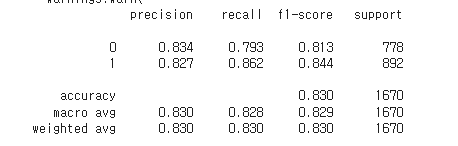
나의 코드는 위의 이미지와 같이 train에 대해서 0.830 정도의 점수가 나왔지만 테스트에서 0.82 정도로 다소 낮은 점수가 나왔다...
데이터 로딩 및 피처 정의
환경은 GPU를 사용하기 위해서 구글 코랩을 사용하였다.
# 1. 데이터 로딩
train = pd.read_csv('/content/drive/MyDrive/train.csv')
test = pd.read_csv('/content/drive/MyDrive/predict_input.csv')
# 2. 피처 정의
feature_cols = [col for col in train.columns if col.startswith(('ecfp_', 'fcfp_', 'ptfp_'))]
meta_cols = ['MolWt', 'clogp', 'sa_score', 'qed']'ecfp', 'fcfp', 'ptfp_' 의 Fingerprint 정보를 별도로 분리하였다.
ecfp_cols = [c for c in feature_cols if c.startswith("ecfp_")]
fcfp_cols = [c for c in feature_cols if c.startswith("fcfp_")]
ptfp_cols = [c for c in feature_cols if c.startswith("ptfp_")]# 3. 학습 데이터 구성
X_train = train[feature_cols + meta_cols].copy()
X_train['ecfp_sum'] = train[ecfp_cols].sum(axis=1)
X_train['fcfp_sum'] = train[fcfp_cols].sum(axis=1)
X_train['ptfp_sum'] = train[ptfp_cols].sum(axis=1)
y_train = train['label']
# 4. 테스트 데이터 구성
X_test = test[feature_cols + meta_cols].copy()
X_test['ecfp_sum'] = test[ecfp_cols].sum(axis=1)
X_test['fcfp_sum'] = test[fcfp_cols].sum(axis=1)
X_test['ptfp_sum'] = test[ptfp_cols].sum(axis=1)여기서 좋은 아이디어라고 생각했던 코드인데, 앞서 EDA과정을 통해 fingerprint 데이터가 중요 feature인 점을 확인하였지만, column의 수가 너무 많아 이를 적용하면 모델의 성능이 저하될 위험이 있었다.
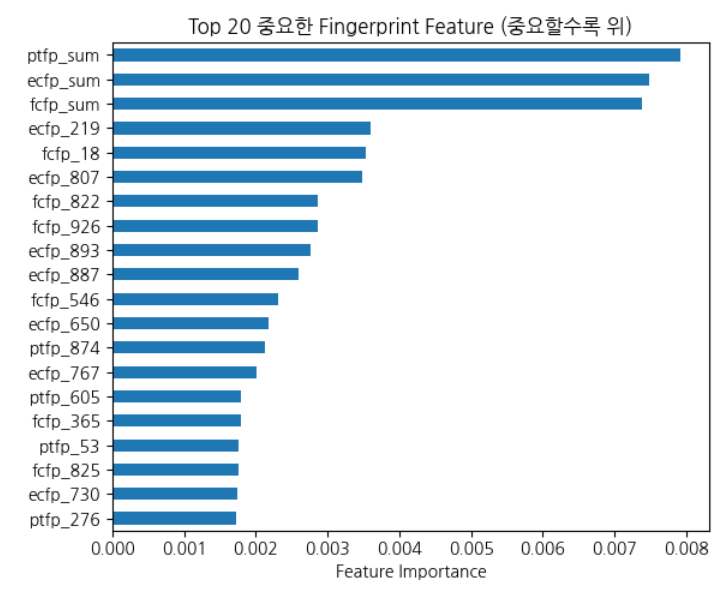
위의 코드와 같이 같은 fingerptint 컬럼끼리의 정보를 합친 파생변수를 만들고, 이에 대하여 randomforest의 importance를 구해보았더니 파생변수의 중요도가 매우 높게 나옴을 확인할 수 있었다.
# 5. Train/Validation 분할
X_train, X_val, y_train, y_val = train_test_split(
X_train, y_train,
test_size=0.2,
stratify=y_train,
random_state=42
)
# 6. SMOTE 오버샘플링
smote = SMOTE(random_state=42)
X_train_over, y_train_over = smote.fit_resample(X_train, y_train)해당 데이터는 8300개 가량의 sample을 가지고 있었고 이는 데이터를 학습시키는 데에 굉장히 부족했다.
따라서 SMOTE 방법론을 활용해서 불균형 데이터에 대해서 데이터를 증강시키는 데에 활용하여 모델의 성능을 높이는 참신한 아이디어였다.
기본적으로도 라벨 1에 대한 score가 높이 나왔는데, 해당 방법을 통해서 confusion matrix에 대해 균형을 맞출 수 있었다

이에 대하여 다음과 같이 모델을 구축하였고,
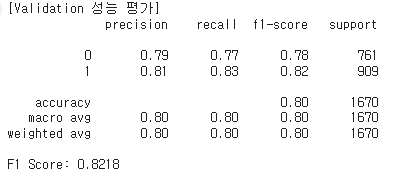
해당 코드는 검증용 데이터에서는 0.8218을 테스트 데이터에서는 0.831에 가까운 좋은 성능을 보였다.

신약 독성 예측~~
주제도 너무 흥미롭네요~~
특히 어떤 사고과정으로 코드를 발전시켰는지 그 과정을 체계적으로 작성해주셨네요~~
이해관계자에 대한 부분만 좀더 develop해주세요~