[Paper Review] Computational methods for RNA modification detection from nanopore direct RNA sequencing data
Computational methods for RNA modification detection from nanopore direct RNA sequencing data.pdf
2021년도 출간
📌 Introduction:
NGS 기술의 주요 한계점:
cDNA를 필요로 하는데 이를 만드는 과정에서 (inosine을 제외한) RNA modification들이 지워짐.
→ (그래서 다음과 같은 방법들과 함께 사용됨.) Antibody-based detection, Chemical-based detection
→ 취약점: i) not quantitative / ii) have high false-positive rates / iii) are inconsistent when using distinct antibodies / iv) do not provide isoform-specific information / v) often lack single nucleotide resolution / vi) require multiple ligation steps and extensive PCR amplification, introducing strong biases in the data
dRNA-seq (direct RNA nonopore sequencing)
ammeter가 ionic current를 k-mer(주로 5-mer)로 측정해 base-calling 알고리즘으로 nucleotide sequence를 알아냄
→ can detect any RNA modification of interest at single nucleotide resolution
→ in individual full-length native RNAs
→ FAST5을 input으로 받고, FASTQ를 output으로 만들어냄.
→ Guppy 가 제일 많이 사용되는데, 4개의 염기만 구분 가능하고 RNA modification과는 아예 무관함. 따라서, 1) FAST5 특징 분석을 통해 RNA modification을 찾아내려고 함. 근데 너무 차이가 적어서 trace나 dwell time 등 추가적인 특징을 더 이용해 성능을 향상시키고 있음; 2) base-calling 과정에서 발생하는 error signature 바탕으로 찾아냄.
📌 Ionic current/signal intensity based methods
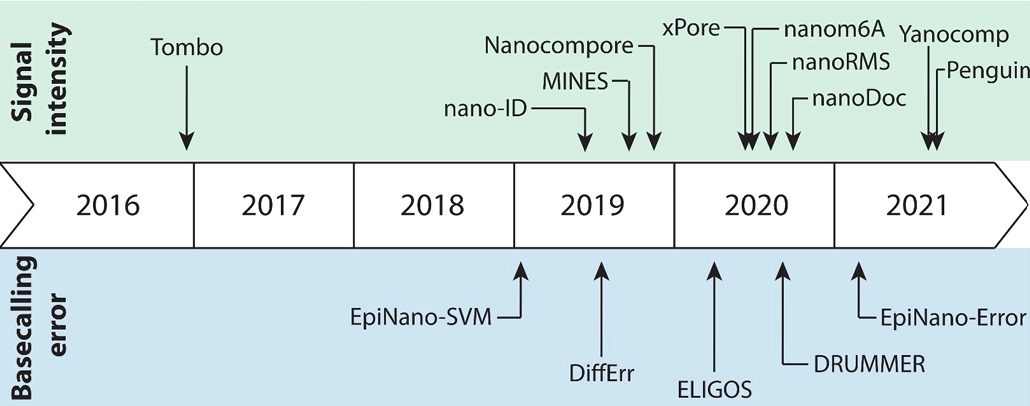
1. Tombo
- minimap2를 이용해서 sequence 를 Reference alignment 함.
- resquiggling (sequence-to-signal assignment):
- signal normalization
- event detection: 전류 패턴이 비교적 안정적인 구간들을 잘라서 이벤트로 만듦.
- sequence-to-signal assignment: 이 event들이 reference sequence의 어떤 k-mer에서 나왔는지 결정.
- modified bases detection (아래 3가지 전략 중 하나 사용)
- using pre-computed models of the signal for specific non-canonical bases
- using a new method that detects any non-canonical base (model X, statistical test O)
- through the comparison with an unmodified sample
2. Nanocompore
- python package
- compared raw electrical signal of sample of interst with sample devoid of RNA modifications (kmer-wise)
- pre-processing steps (input data)
- base-calling
- mapping to the reference transcriptome
- sequence-to-signal assignment (resquiggling) (by Nanopolish)
- feature 생성 (detect differences)
- each kmer of each read의 signal intensity의 median(중앙값), dwell time(나노포어 체류 시간) 계산
- grouping reads → analysing with a statistical test (GMM) → logistic regression 또는 signal intensity나 dwell time 중 한 가지만을 이용한 모수적 / 비모수적 단변량 통계 검정
- GMM: 모든 샘플의 median signal intensity와 dwell time
3. xPore
- python package (Nanocompore랑 비슷함)
- Nanopolish로 sequence에 재정렬된 signals를 사용, GMM 이용해서 modified sites를 찾아냄.
4. NanoRMS
5. Nanom6A
6. MINES
7. nanoDoc
8. Penguin
9. Yanocomp
10. Nano-ID
📌 Base-calling and alignment-based methods
RNA modification은 ionic current를 변화시켜 base-calling 과정에서 specific한 site에 mismatch와 indel이 반복적으로 축적되도록 한다.
따라서, RNA modification를 검출하기 위해 error frequency를 background와 비교하는 방법들이 개발되었으며, background는 알고리즘 훈련용으로 사전 정의되거나 paird 실험 설계(modification이 있는 조건과, 거의 없는 조건을 한 쌍으로 비교하는 실험 설계)를 통해 구축될 수 있다.
이러한 접근 방식은 multiple reads를 기반으로 modified sites의 위치를 제공하지만 single-molecule resolution은 제공하지 않는다.
1. EpiNano
- Version 1.0
- Support Vector Machines(SVMs)를 사용해 m6A-modified sites를 예측함.
- Knock out condition을 필요로 하지 않고, 이전 m6A modifed data들을 이용해서 훈련함.
- paired conditions(e.g. wild type and KO)에서 사용하기를 권장
- Version 1.2와 그 이후
- EpiNano-SVM
- pre-trained SVMs를 이용함
- EpiNano-Error
- 어떤 RNA modification도 예측할 수 있음.
- pre-trained SVM models를 필요로 하지 않음.
- 비교할 paired conditions를 필요로 함.
2. DiffErr
- WT와 KD을 비교해 m6A sites를 찾음
- G-tests to a 2 x 5 contingency table
| A | C | G | U | indel | |
|---|---|---|---|---|---|
| 각 샘플 |
- P-value < 0.05 인 site에 대해서, 같은 조건의 replicates와 비교하기 위해 두번째 G-tests를 수행함.
- 만약 replicates 사이의 G statistic이 WT와 KD 사이의 G statistic보다 크다면, 해당 위치는 조건 차이보다 실험 잡음(noise)이 더 크다고 판단해 제거함.
- m6A 판정 조건
1) Benjamini-Hochberg 보정 P-value < 0.05
2) log fold change in mismatch to match ratio between WT and KD >1
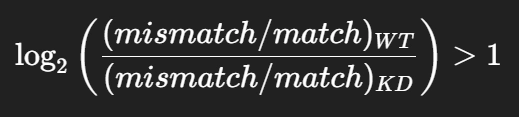
*G 통계량 (G-test): 두 분포가 얼마나 다른지를 수치로 표현한 값
- 값이 크다 → 두 분포가 많이 다름
- 값이 작다 → 두 분포가 비슷함
- p < 0.05
- 이런 차이가 우연히 나올 확률 < 5%
- 우연이라고 보기엔 너무 드물다
3. DRUMMER
- DiffErr와 상당히 유사함.
- m6A 판정 조건
1) Bonferroni 보정 P-value < 0.01
2) WT sample의 (matches/mismatches) 값 ≤ 1/2 x KD sample의 (matches/mismatches) 값
4. ELIGOS
- matches와 mismatches (i.e. substitutions, insertions, deletions)를 비교해 modified sites를 찾음.
- reference sequence를 정답으로 두고, 5-mer에서 mismatch frequency 계산함
- 해당 염기와 앞 염기, 뒤 염기까지 테스트 수행해 P-value 계산해 Benjamini–Hochberg로 보정함. 각 염기에서 얻은 최대 odds ratio를 최종 결과로 사용함.
*IVT(In Vitro Transcription): 시험관(in vitro)에서 합성한 RNA로, 완전히 통제된 조건에서 생성됨. RNA modification 효소(writer)가 없으므로 unmodified 상태임.
*de novo: 처음부터, 새로이
*per se: 그 자체로, 본질적으로
