✅ CADD (Computer Aided Drug Discovery)
CADD란 Lead identification 단계를 효율적으로 진행하기 위해 컴퓨터를 이용하는 것을 말한다.
](https://velog.velcdn.com/images/ssumannb/post/0c8f79e1-edab-4110-810b-3846689697b8/image.png) https://bioinformaticsandme.tistory.com/26
https://bioinformaticsandme.tistory.com/26
CADD의 접근은 물질(ligand)이 작용하기 위해 자신에게 맞는 수용체(receptor)에 결합한다는 개념으로부터 출발한다.
🔴 Structure based Drug Design
receptor의 구조에 따라 약물을 디자인하는 개념
🔴 Ligand-based Drug Design
Receptor에 결합하는 ligand에 초점을 맞춰 디자인 하는 개념
✅ Structure Based Drug Design
표적 단백질의 구조를 기반으로 하여 거기에 가장 잘 결합할 수 있는 물질을 디자인 하는 과정이다.
1) 먼저 표적의 구조를 밝히고, 2) 표적에 물질이 어떻게 결합할 것인지를 예측한 뒤, 3) 가장 잘 결합할 수 있는 순서대로 랭킹을 매기는 방식으로 진행된다.
1. Protein Structure Determination (단백질 구조 결정)
표적 단백질의 3D 구조를 확인하는 단계이다
Experimental Method (실험적 방법)
단백질의 구조를 결정하기 위해 직접 실험하는 것. 구조가 밝혀진 단백질은 보통 DB나 library에 등록되어 있다.
- X-ray Crystallography (X선 결정학): X선의 회절을 이용하여 결정의 원자와 분자 구조를 밝혀내는것을 말한다. 결정학으로 결정 내의 전자 밀도의 3차원 그림을 만들어낼 수 있다.
- Nuclear Magnetic Resonance (핵자기공명법; NMR):
위 두 방법이 대표적인 실험을 통한 단백질 구조 결정 방법이다.
Homology Modeling (상동성 모델링)
비교 모델링 방법(comparative modeling)이라고도 한다.
단백질의 아미노산 시퀀스로부터 원자단위의 모델을 구성하기 위해 참고하는 모델링 방법으로, 유사한 아미노산 서열을 가진 단백질들은 서로 유사한 3차원 구조를 가질 것이라는 가정에서 출발한다.
대상 단백질의 아미노산 서열과 유사한 서열을 가지는 단백질(template: 주형)이 PDB 내에 존재할 경우 (즉, 3차원 구조가 알려졌을 때), 주형 단백질의 구조를 이용하여 대상 단백질의 구조를 계산하는 방법이다.
Fold Recognition (구조 인식 방법)
Threading이라고도 한다.
주어진 아미노산 서열에 따라 단백질이 어떻게 접힐지를 예측함으로써 구조를 알아내는 방법
New Fold Method (새 구조 방법)
2. Docking
약물이 목표로 하는 표적에 어떻게 결합할 것인지를 뜻한다.
Def. "Docking is an automated computer algorithm, that determines how a compound will bind in the active site of a protein"
일반적으로 docking을 사용하는 경우 Binding site를 지정해주고 지정된 범위 안에서 ligand의 conformation을 찾는다. Ligand가 결합된 상태로 구조가 규명되어 있는 경우 쉽게 binding site를 찾울 수 있지만, 그렇지 않은 경우는 표적 단백질과 비슷한 기능을 하는 단백질 family등에서 비교하여 찾거나, binding site 예측 프로그램을 사용한다.
*Binding site에 대한 정보를 주지 않고 docking을 진행하는 경우는 blind docking이라고 한다.
💡 Ligand conformation : ligand 형태 (흔히 pose라고 함)
Binding affinity : 결합 친화성 (scoring 함수를 통해 평가한다)
방식에 따른 분류 1- Rigid
방식에 따른 분류 2- Semi-flexible
방식에 따른 분류 3- flexible
Stochastic-based algorithm
Stochastic 탐색 알고리즘은 ligand conformation을 임의로 수정하면서 가능한 conformation을 찾아가는 방법으로, 임의로 ligand를 회전시키거나 이동시키면서 다양한 pose를 만든다. 만들어진 pose들은 에너지 값 혹은 scoring 함수를 기반으로 선택된다. 선택된 conformation 만을 이용하여 다음 단계 pose를 생성하는 과정을 반복해서 최종적으로 pose를 제시하게 된다.
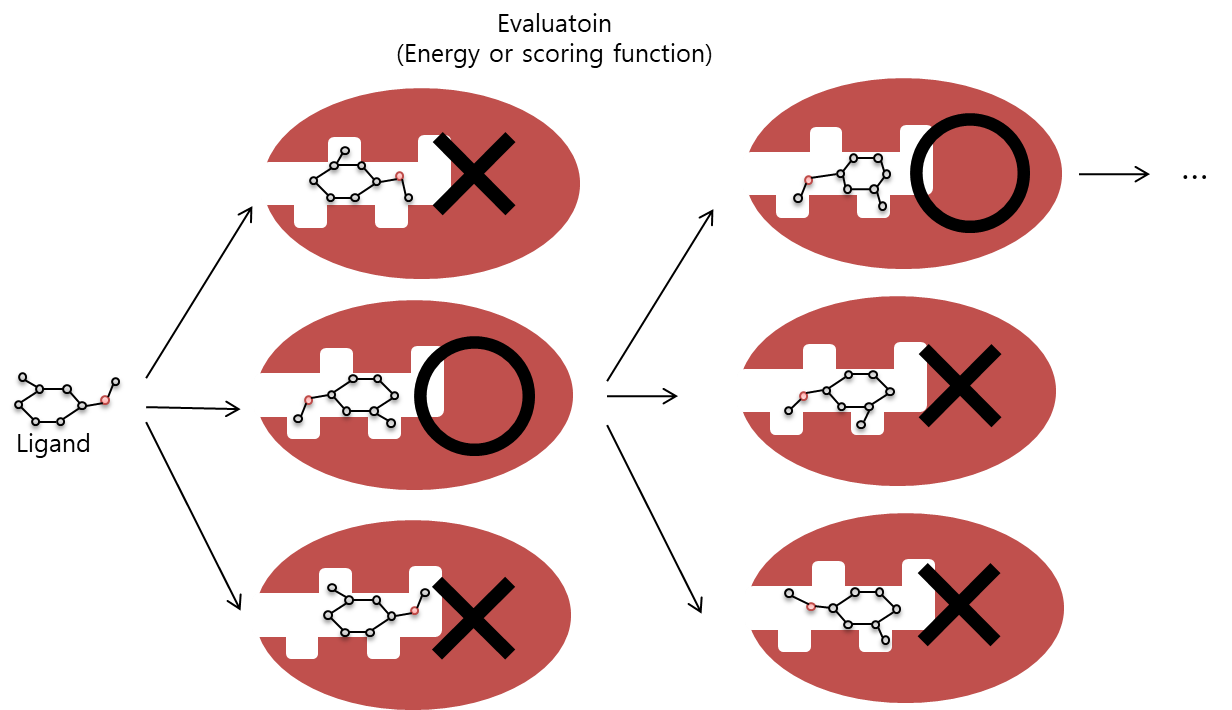
https://www.ibric.org/myboard/read.php?id=291753&Board=news
Systemic
Deterministic
Pharmacophore-based algorithm
* Pharmacophore : 약물이 활성을 나타내는데 중요한 부분들
- 분자의 형태와 화학적 정보를 기반으로 매칭시키는 방법이다.
- 단백질의 active site와 ligand를 pharmacophore로 표현해서 각 분자별 pharmacophore간의 거리를 기반으로 매칭이 이루어지는 알고리즘이다.
- 각 분자별 pharmacophore 간의 거리를 기반으로 매칭이 이루어진다.
- 새로운 conformation을 갖는 ligand를 생성할 때 pharmacophore간의 거리 및 pharmacophore에 해당하는 원자를 기반으로 구조를 만들게 되며, 수소 결합(hydrogen-bond) donor 혹은 acceptor와 같은 화학적인 정보를 반영하여 매칭을 진행할 수 있다.
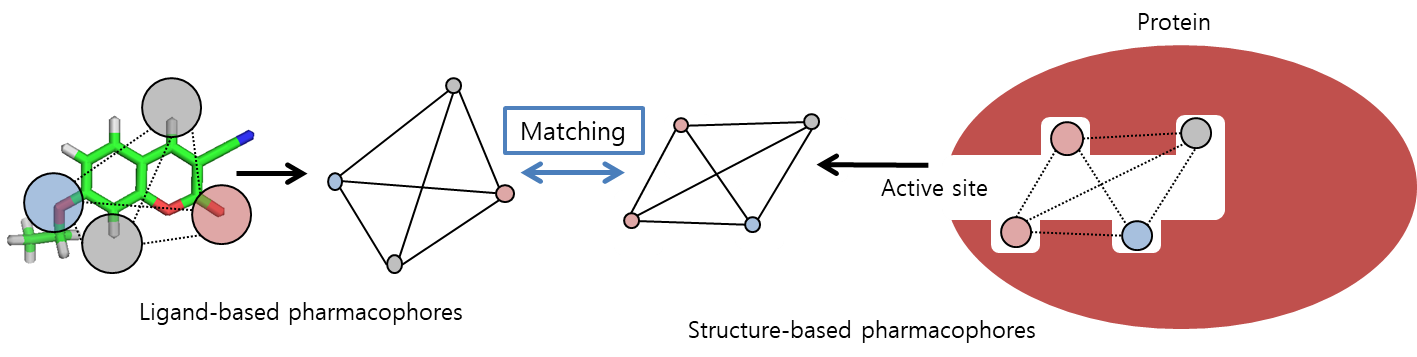
Fragment-based algorithm
Ligand를 몇몇 조각(fragment)들로 나눈 후 각각의 조각들을 docking하는 방법이다.
조각들 중 가장 큰 조각을 먼저 docking 시키는 데 이는 가장 큰 조각이 단백질과의 상호작용에 가장 중요한 기능을 할 것으로 생각되기 때문이다. 큰 조각이 docking 된 후 그 조각에 fragment들을 차례로 추가시키면서 pose를 만든다
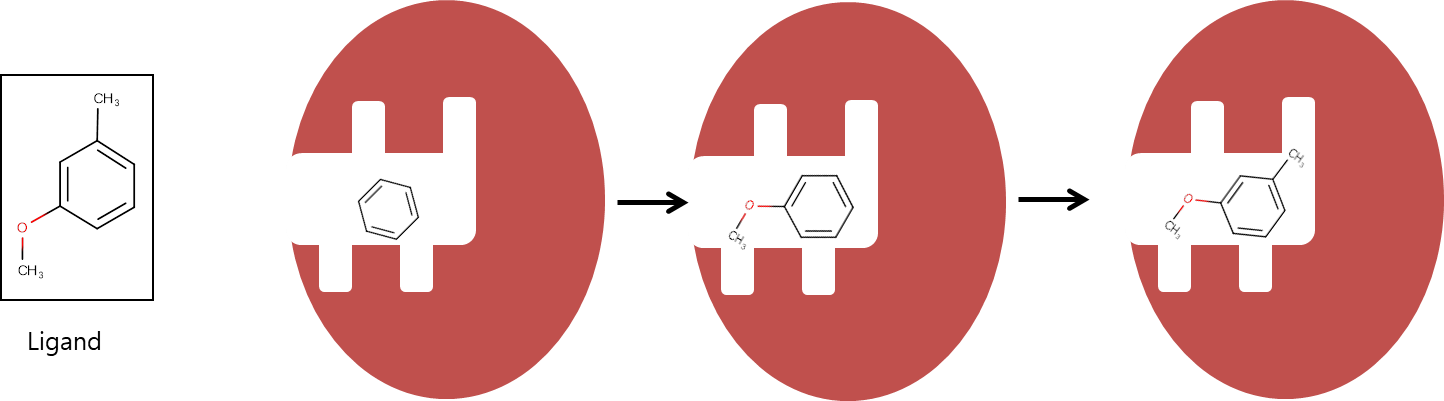
3. Binding Free Energy (Scoring)
Docking 까지 예측했다면, 어떤 물질이 잘 결합할 것인지도 예측하여 후보물질의 순위를 매길 수 있다.
물질이 표적에 결합할 때 protein-ligand interaction이 일어날 때 발생하는 binding free energy 개볌이 이용된다. 이 에너지가 의미하는 것은 물질과 타겟의 궁합이다.
✅ Ligand Based Drug Discovery
표적 단백질의 구조가 밝혀져 있지 않거나, 각종 구조 예측 모델링으로도 예측이 불가능 할 때에는, 관점을 바꾸어 표적에 결합하는 물질인 ligand의 관점에서 접근한다.
computer 모델을 이용하여 다양한 변수들을 바꿔가며, 구조를 모르는 표적에 가장 잘 결합할 수 있을 것 같은 물질을 찾아내는 방법이다.
🟡 QSAR (Quantitative Structure-Activity Relationship)
LBDD에서 가장 널리 쓰이는 방식
Ligand의 구조가 변함에 따라 표적 결합 시 활성화 정도가 어떻게 변하는지를 수치화 하는 것으로 이해할 수 있다.
🟡 3D-QSAR (3-Dimentional Quantitative Structure-Activity Relationship)
[Initiation] 인공지능과 신약개발: Application of Artificial Intelligence to Drug Discovery
