왜 생겼고, 왜 반드시 거쳐야 하는지
HPC를 쓰기 시작하면 아무리 늦어도 결국 마주치게 되는 게 Slurm이다. 처음엔 그냥 “GPU 쓰려면 이거 해야 한대” 수준으로 지나가지만, 조금만 써보면 깨닫게 된다. Slurm은 선택지가 아니라 전제 조건이라는 것을...

이미지 출처 : 위키백과
1. Slurm은 왜 필요해졌을까
GPU / CPU는 항상 부족하다
연구실에 사람이 100명 있고 GPU가 16개면, 누가 지금 GPU를 쓰는지 알 수 없고, 먼저 접속한 사람이 계속 점유하고, 누군가 실수로 프로세스 날리고, 학습 중이던 실험은 그대로 터진다. 이게 “이론적으로”가 아니라 실제로 흔하게 생긴다. 그리고 공용 서버는 이런 상황이 한 번 터지면 진짜로 다 같이 피곤해진다.
공정하게 나눌 방법이 필요했다
딥러닝 학습은 몇 분짜리 작업이 아니라 보통 수시간~수십시간 단위고, 길면 며칠 동안 GPU를 붙잡는다. 이걸 아무 제약 없이 풀어두면 서버는 곧 몇 명의 전유물이 된다. 그래서 “누가, 어떤 자원을, 얼마나 쓰는지”를 시스템이 강제로 관리하는 구조가 필요해졌고, 그 역할을 하는 게 스케줄러다.
자동화 없이는 운영이 안 된다
사람 손으로는 절대 운영이 안 된다. job 끝나면 다음 작업 자동 실행, 자원 사용 기록, 여러 노드에 걸친 분산 작업 실행 같은 걸 사람이 매번 수동으로 한다? 불가능하다. 그래서 Job Scheduler가 등장했고, HPC 환경에서는 Slurm이 사실상 표준처럼 굴러간다(센터/기관마다 PBS 같은 다른 스케줄러도 있긴 한데, 국내 대학/연구실은 Slurm 비율이 진짜 높다).
2. Slurm의 기본 사고방식
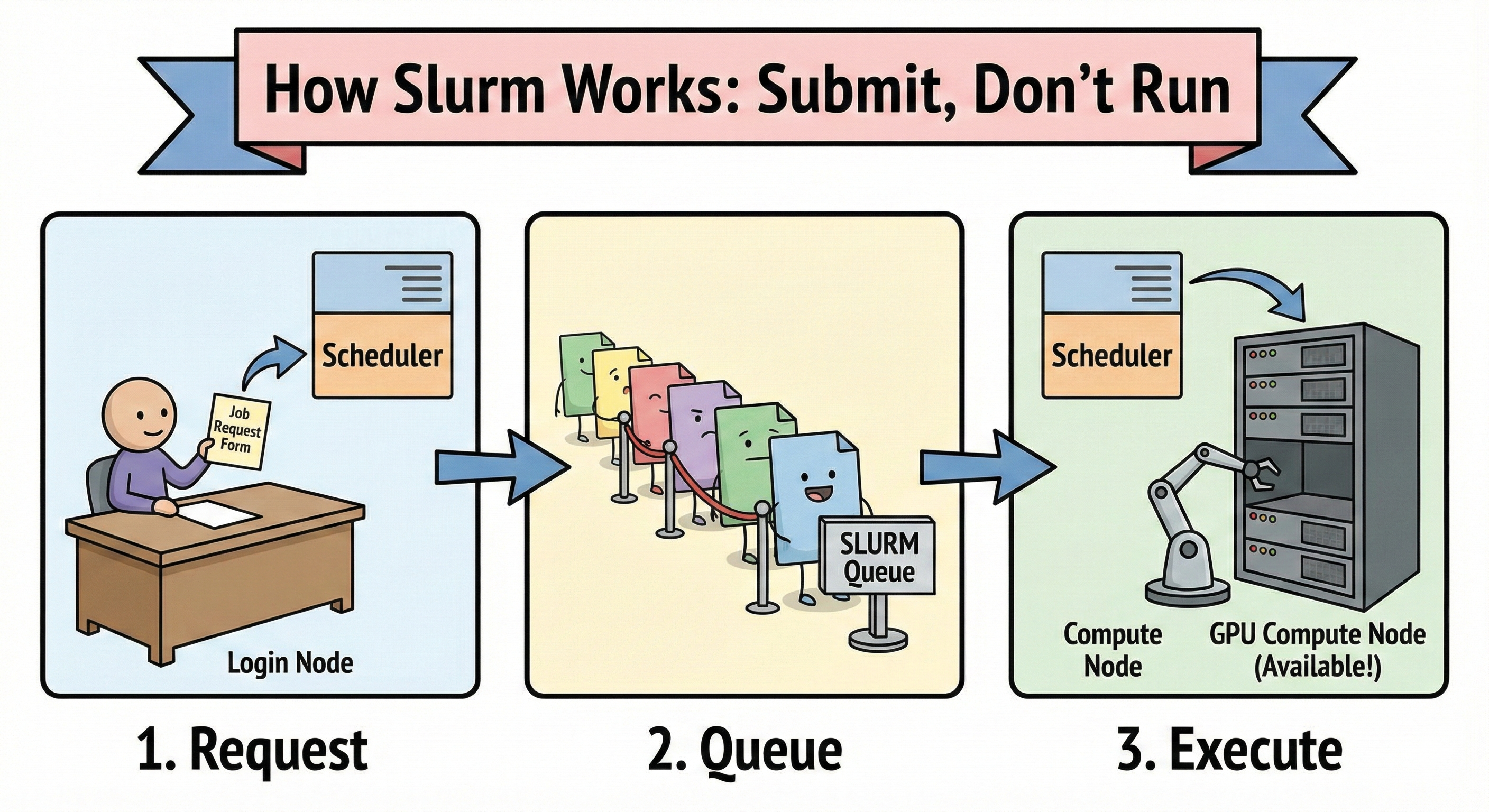
복잡하게 생각할 필요 없다. Slurm이 하는 일은 딱 세 가지다. 자원을 예약하고, 작업을 큐(queue)에 넣고, 빈 자리가 나면 알아서 실행한다. 즉 “GPU를 직접 잡고 쓰는 것”이 아니라 GPU 사용권을 요청해서 배정받는 구조라는 게 핵심이다. 여기서 제일 중요한 마인드셋은 이거다: 로그인 노드에서 뭔가를 돌리는 게 아니라, 계산 노드에서 돌릴 수 있게 ‘요청서를 제출하는 방식’이다.
3. HPC에서의 기본 흐름
항상 이 순서로 돌아간다.
User → Login Node → Slurm Scheduler → Compute Node (GPU/CPU)
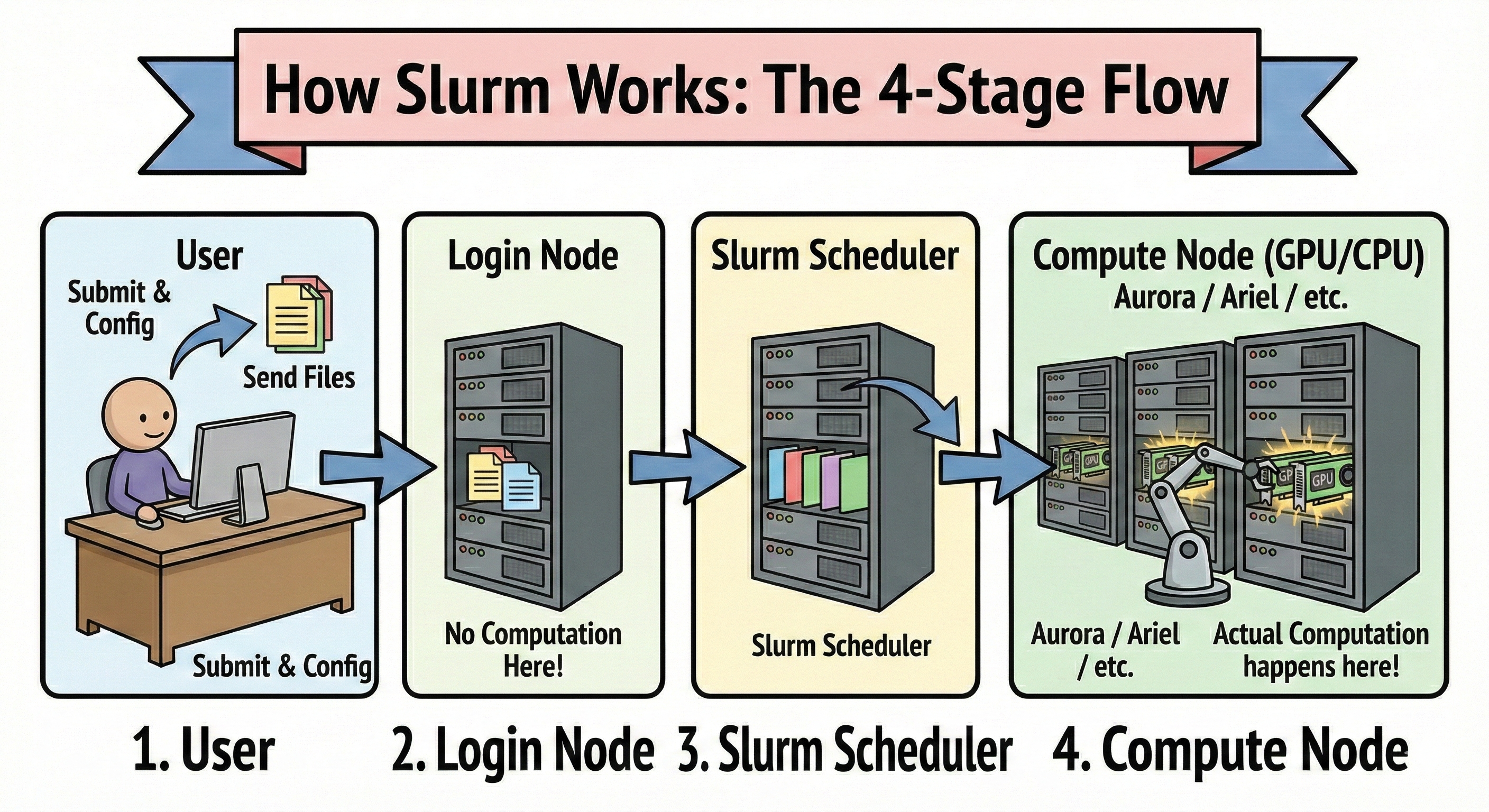
로그인 노드는 접속해서 파일 올리고, 환경 잡고, 작업 제출하는 곳이다. 여기서 학습을 돌리면 안 된다(정확히는 “절대 금지”로 막아둔 곳도 많고, 안 막아도 하면 민폐가 된다). 스케줄러(Slurm)는 누가 뭘 요청했는지 보고 순서랑 자원 배치를 결정한다. 계산 노드는 실제 GPU 연산이 일어나는 곳이다. Aurora든 Ariel이든 큰 구조는 거의 비슷하고, 달라지는 건 “GPU 종류/노드 스펙/파티션 정책/시간 제한” 같은 운영 디테일이다.
4. 가장 많이 쓰는 Slurm 명령어들
외울 필요는 없고 자주 쓰는 것만 손에 익히면 된다(고 생각하긴한다)
| 목적 | 명령어 |
|---|---|
| 클러스터 상태 | sinfo |
| 내 job 보기 | squeue -u $USER |
| 전체 job 보기 | squeue |
| job 제출 | sbatch run.sh |
| 인터랙티브 실행 | srun --pty bash |
| 인터랙티브 GPU | srun --gres=gpu:1 --pty bash |
| job 취소 | scancel JOBID |
| job 통계/자원 사용량 | sacct -j JOBID |
| GPU 상태(노드 안에서) | nvidia-smi |
| 로그 확인 | tail -f logs/xxx.out |
여기서 포인트 하나. nvidia-smi는 “로그인 노드에서 치는 명령”이 아니라, GPU가 잡힌 계산 노드에서 확인하는 용도다. 그래서 보통 srun으로 노드 들어간 다음에 확인하는 흐름이 자연스럽다.
그리고 sinfo 봤을 때 파티션(partition)이 여러 개면, 그건 “GPU용 큐 / CPU용 큐 / 수업용 큐 / 연구용 큐” 이런 식으로 나뉘어 있는 경우가 많다. 그때는 --partition=... 옵션을 써야 할 때가 있다(안 쓰면 기본 파티션으로 들어가거나, 아예 제출이 거절될 수도 있음).
5. sbatch vs srun (처음에 제일 헷갈리는 포인트)
sbatch
배치 작업이다. 스크립트 제출하면 큐에 들어가고, 차례가 오면 알아서 시작되고, 알아서 끝난다. 결과는 로그 파일로 확인한다. 긴 학습, 실험 돌리기, 밤새 돌리기 전부 sbatch다.
srun
인터랙티브 작업이다. GPU 붙은 쉘을 하나 받는 느낌이다. 디버깅, 환경 확인, “이 코드 돌아가나?” 테스트할 때 쓴다. 단, 오래 돌릴 거면 srun으로 버티는 게 아니라 sbatch로 넘기는 게 정석이다(연결 끊기거나, 세션 꼬이면 피곤해진다).
정리하면 이거다. “이 코드가 돌아갈까?” → srun / “이제 제대로 학습 돌린다” → sbatch
6. 최소 Slurm 스크립트 예시
#!/bin/bash
#SBATCH --job-name=train
#SBATCH --gres=gpu:1
#SBATCH --time=24:00:00
#SBATCH --output=logs/%j.out
source ~/.bashrc
conda activate myenv
python train.py \
--epochs 3 \
--batch_size 16여기서 %j는 job id로 자동 치환된다. 로그는 무조건 파일로 남기는 게 맞고, stdout만 믿고 nohup 같은 걸로 버티는 건 HPC에서는 굳이 추천하지 않는다(어차피 Slurm이 로그를 남겨준다). 그리고 은근히 중요한 게 하나 더 있는데, logs/ 폴더는 미리 만들어놔야 한다. 없으면 출력 경로 때문에 job이 바로 실패하는 경우도 있다.
실제로는 여기서 메모리/CPU도 같이 지정하는 경우가 많다. 예를 들면 --cpus-per-task=4, --mem=16G 같은 옵션들. GPU만 잡아놓고 CPU나 메모리를 너무 적게 잡으면 데이터 로더에서 병목 걸리거나, 반대로 메모리 부족으로 학습이 터질 수도 있다. 그러니까 “GPU만 있으면 끝”이 아니라, GPU+CPU+RAM이 세트로 돌아간다고 생각하는 게 편하다.
결론
Slurm은 GPU를 쓰기 어렵게 만드는 시스템이 아니라, 여러 사람이 안전하게 쓰게 만드는 시스템이다. 처음엔 귀찮고 명령어도 많아 보이지만, sinfo, squeue, srun, sbatch 이 네 개만 익숙해지면 HPC의 절반은 넘은 거다. 그리고 진짜 중요한 건, “내가 지금 어느 노드에서 뭘 돌리고 있는지” 이걸 계속 의식하는 습관이다. 로그인 노드에서 돌리는 순간부터 대부분의 사고가 시작한다.
