Convoluted neural networks (CNN)에 대해 배웠습니다. 왜 많은 사람들이 computer vision영역에서 일하고 싶어하는지 알 것 같습니다, 너무 신기하고 재밌어요..!
컴퓨터에게 시각정보처리 능력을 주기에 CNN의 활용방법은 무궁무진하지만, CNN을 처음 배우는 사람들은 모두 인간이 손으로 쓴 숫자 (handwritten digits)를 알아볼 수 있는 인공신경망을 구현하는 연습을 합니다 (MNIST 데이터셋을 사용해서요).
그와 비슷하면서도 초보자가 연습하는데 써볼 수 있을만한 데이터셋이 없을까 하고 인터넷을 찾아보다가, 적절한 데이터셋을 찾았습니다. 미국 National Institute of Health의 말라리아 데이터셋입니다 (두 번째 링크 Kaggle에 올라온 버전이 활용이 더 쉽습니다):
Malaria Datasheet (nih.gov)
Malaria Cell Images Dataset | Kaggle
말라리아 개론
말라리아에 대한 간단한 소개- Plasmodium이라는 기생충이 인간의 적혈구 속에 들어가서 기생하는 병입니다 (주로 모기에 의해 전파됩니다)
- 전 세계적으로 매년 5억명 정도 걸린다고 합니다 (어마어마하죠..? 지구의 역사상 태어난 모든 인간의 절반이 말라리아에 의해 죽었을 수 있다는 견해도 있습니다 [1] 현대의학의 발전으로 선진국에서는 먹는 약으로 비교적 쉽게 치료가 가능하지만, 개발도상국들에선 아직도 사망 원인 10위 안에 든다고 합니다 [2])
- 우리나라에도 말라리아 있습니다 (경기, 강원 북부에 특히)
- 말라리아는 적혈구에 사는 기생충이 있는 병이기 때문에, 진단 방법은 간단합니다: 혈액을 현미경으로 봐서 적혈구에 기생충이 있는지 없는지를 보면 됩니다
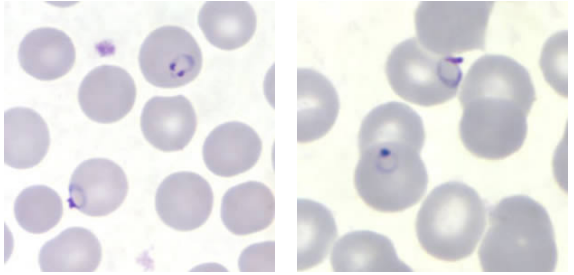
(Source: CDC, Pfalciparum_benchaidV2.pub (cdc.gov))
위 사진은 현미경으로 본 적혈구들이고, 몇몇 적혈구 안에 있는 반지처럼 생긴 것들이 바로 Plasmodium 기생충입니다 (생장단계에 따라 다른 모습으로 나타날 수 있지만, 이 반지 형태로 관찰되는 경우가 가장 흔한 것으로 알고 있습니다). 이런 기생충이 있는 적혈구들이 관찰되면 말라리아로 진단이 가능한 것이죠.
NIH의 말라리아 데이터셋은 정상적혈구들과 말라리아 기생충이 있는 적혈구들의 이미지 데이터셋입니다:
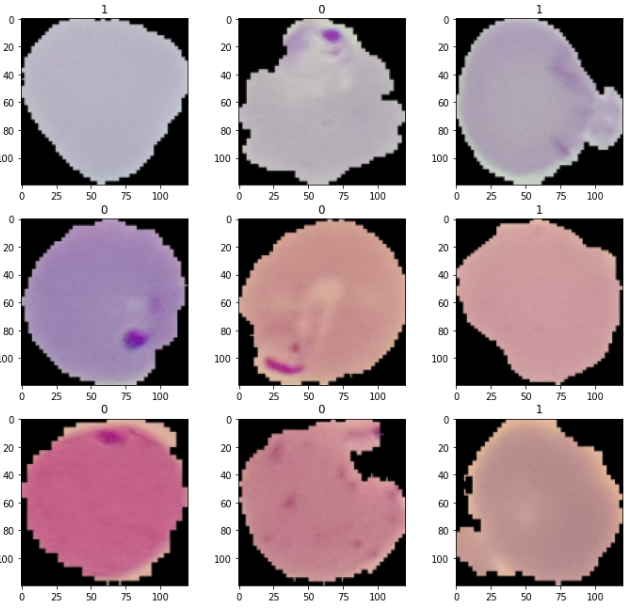
(Visualized from NIH's Malaria Data (Thin Smears - Falciparum and uninfected patietns) Malaria Datasheet (nih.gov))
어떤 적혈구들이 말라리아에 감염 된 것들이고, 어떤 적혈구들이 정상인지 알아보실 수 있겠나요?
사실 그렇게 어렵지 않게 구분 가능한 것 같습니다. 이미지 위에 '1'이라고 되어있는 것들은 정상 적혈구들이고 '0'이라고 되어 있는 것들은 말라리아 적혈구들입니다.
말라리아 잡는 CNN 만들어보기
각 혈구를 이미지로 만들어 놓아서 데이터가 이미 너무 깔끔한 상태라 바로 말라리아가 있는 적혈구와 정상 적혈구를 구분하는 CNN을 구현해볼 수 있습니다. 저는 Google Colab을 이용했습니다.import numpy as np
import pandas as pd
import matplotlib.pyplot as plt
%matplotlib inline
import os
import cv2
from PIL import Image
from torch.utils.data import Dataset, DataLoader
import torch
import torch.nn as nn
from torchvision import transforms, datasets, models이미지 데이터를 활용하는 tip:
이미지 데이터가 들어있는 폴더는 용량이 크기 때문에 보통 zip파일로 접하게 됩니다. Colab에서는 다음 커멘드를 실행하여 zip파일을 쉽게 활용할 수 있습니다:
!unzip drive/MyDrive/...zip파일위치.../...zip파일이름.zip이렇게 생성 된 이미지 폴더가 있으면 torchvision의 datasets.ImageFolder를 이용해 Dataset을 만들어줄 수 있습니다. 이 때 이미지 데이터를 모두 120 x 120 pixel로 리사이징하고 (이미지 사이즈가 모두 동일하지 않으면 뒤에 DataLoader로 각 이미지에 대해 iteration하며 모델을 트레이닝할 때 에러가 발생하더라구요) pytorch의 tensor로 변환해서 읽어오도록 하겠습니다.
# Define data transformations to be performed when reading in the images
data_transforms = transforms.Compose([transforms.Resize((120, 120)),
transforms.ToTensor()])
# Location of images
img_dir = '/content/cell_images/cell_images'
malariadata = datasets.ImageFolder(img_dir, transform=data_transforms)datasets.ImageFolder는 각 이미지가 들어있는 폴더를 그 이미지의 label(즉 class)로 인식합니다. datasets.ImageFolder를 통해 읽어 온 데이터의 label들을 보면
print(malariadata.class_to_idx)
Parasitized (말라리아에 감염 된 적혈구 이미지들)이 하나의 label, Uninfected (정상 적혈구 이미지들)이 하나의 label을 구성하고 있는 것을 확인할 수 있습니다.
다음으론 이미지들을 training set과 test set으로 나눠보겠습니다 (Train test split하는 방법은 여러 가지기 때문에 꼭 이 방법으로 할 필욘 없습니다)
from torch.utils.data.sampler import SubsetRandomSampler
test_size = 0.2
data_length = len(malariadata)
indices = list(range(data_length))
np.random.shuffle(indices)
test_split = int(np.floor(test_size*data_length))
test_index, train_index = indices[:test_split-1], indices[test_split:]
train_sampler = SubsetRandomSampler(train_index)
test_sampler = SubsetRandomSampler(test_index)
train_loader = DataLoader(malariadata, sampler=train_sampler, batch_size=32)
test_loader = DataLoader(malariadata, sampler=test_smapler, batch_size=32)(Batch size를 어떻게 정하는게 가장 좋은지는 잘 모르겠습니다. 곧 소개할 논문을 따라서 32로 정했습니다.)
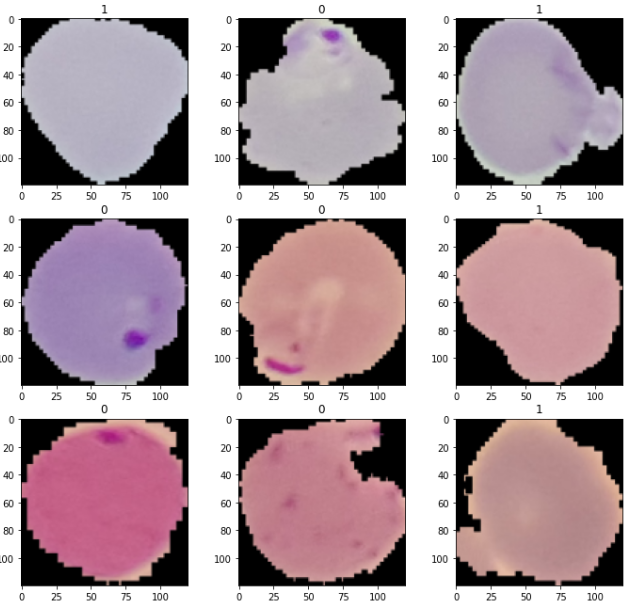
train_loader를 통해 이미지 몇 장을 확인해보도록 하겠습니다:
img_tensors, labels = next(iter(train_loader))
def showimg(img_tensor):
#use matplotlib to display an image that is in tensor form
npimg = img_tensor.numpy()
plt.imshow(np.transpose(npimg, (1,2,0)))
fig = plt.figure(figsize=(20,15))
for i in range(20):
ax = fig.add_subplot(4, 5, i+1, title=labels[i].item())
showimg(img_tensors[i])
plt.show()
(Visualized from NIH's Malaria Data (Thin Smears - Falciparum and uninfected patietns) Malaria Datasheet (nih.gov))
그럼 이제 활용할 CNN을 정의해보도록 하겠습니다. Umer et al의 "A Novel Stacked CNN for Malarial Parasite Detection in Thin Blood Smear Images" (IEEE Xplore Full-Text PDF:)[3] 논문에 나온 CNN 구조를 따라 만들어보려 했습니다. 그런데 논문에서 코드는 따로 공개하고 있지 않고, kernel의 padding이나 stride 등 몇 가지 디테일은 명시하고 있지 않아서 그런 부분은 제 마음대로 넣고 만들어보았습니다. 논문의 Figure4가 CNN 구조를 한 눈에 보여줍니다 (여담: 한 1주일 전만해도 이런 그림보면 멋있긴한데 뭔뜻이지 싶었는데 이제 이해가 가서 너무 기쁩니다).
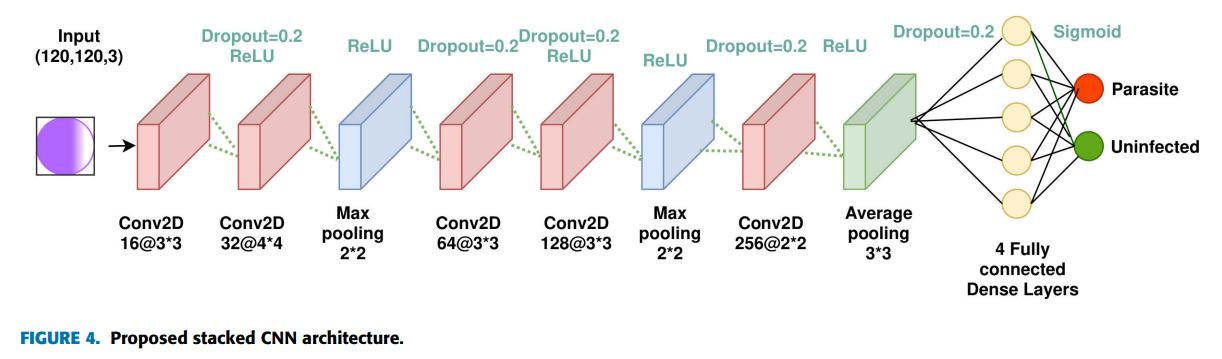
(Source: IEEE Access Vol 8 2020. Umer et al [3], Figure 4. CC-BY-4.0)
Pytorch로 다음과 같이 재현해봤습니다 (뒤쪽에 fully connected layer 단계에서 저자들은 activation function으로 sigmoid function을 사용하는데, 저는 그냥 익숙한 ReLU를 썼습니다):
# Recreation of CNN described by
# https://ieeexplore.ieee.org/stamp/stamp.jsp?arnumber=9093853
class MalariaNet(nn.Module):
def __init__(self):
super().__init__()
self.layer1 = nn.Sequential(
nn.Conv2d(3, 16, kernel_size=3, stride=1, padding=1),
nn.Dropout2d(0.2),
nn.ReLU(),
nn.Conv2d(16, 32, kernel_size=4, stride=1, padding=2),
nn.ReLU(),
nn.MaxPool2d(kernel_size=2, stride=2)
)
self.layer2 = nn.Sequential(
nn.Conv2d(32, 64, kernel_size=3, stride=1, padding=1),
nn.Dropout2d(0.2),
nn.ReLU(),
nn.Conv2d(64, 128, kernel_size=3, stride=1, padding=1),
nn.ReLU(),
nn.MaxPool2d(kernel_size=2, stride=2)
)
self.layer3 = nn.Sequential(
nn.Conv2d(128, 256, kernel_size=2, stride=1, padding=1),
nn.ReLU(),
nn.AvgPool2d(kernel_size=3, stride=3)
)
self.fc = nn.Sequential(
nn.Linear(256*10*10, 512),
nn.ReLU(),
nn.Linear(512, 256),
nn.ReLU(),
nn.Linear(256, 128),
nn.ReLU(),
nn.Linear(128, 2)
)
self.fla = nn.Flatten()
self.drop = nn.Dropout2d(0.2)
def forward(self, x):
out = self.layer1(x)
out = self.drop(out)
out = self.layer2(out)
out = self.drop(out)
out = self.layer3(out)
out = self.drop(out)
out = self.fla(out)
out = self.fc(out)
return out이제 MalariaNet 클래스로 모델을 만들어서 트레이닝 시켜보겠습니다:
device = torch.device("cuda" if torch.cuda.is_available() else "cpu")
model = MalariaNet()
model.to(device)
error = nn.CrossEntropyLoss()
optimizer = torch.optim.Adam(model.parameters(), lr=0.001)
# Train the model
num_epochs = 30
for epoch in range(num_epochs):
train_loss, train_acc = 0, 0
model.train() # state that model training is beginning
for (images, labels) in train_loader:
images, labels = images.to(device), labels.to(device)
predictions = model(images)
optimizer.zero_grad()
loss = error(predictions, labels)
loss.backward()
optimizer.step()
train_loss += loss.to('cpu').item()
train_acc += (predictions.argmax(1)==labels).type(torch.float).to('cpu').mean().item()
train_loss /= len(train_loader) #len(train_loader) is batch size
train_acc /= len(train_loader)
print(f"Epoch: {epoch}, loss: {train_loss:>6f}, acc: {train_acc:>6f}")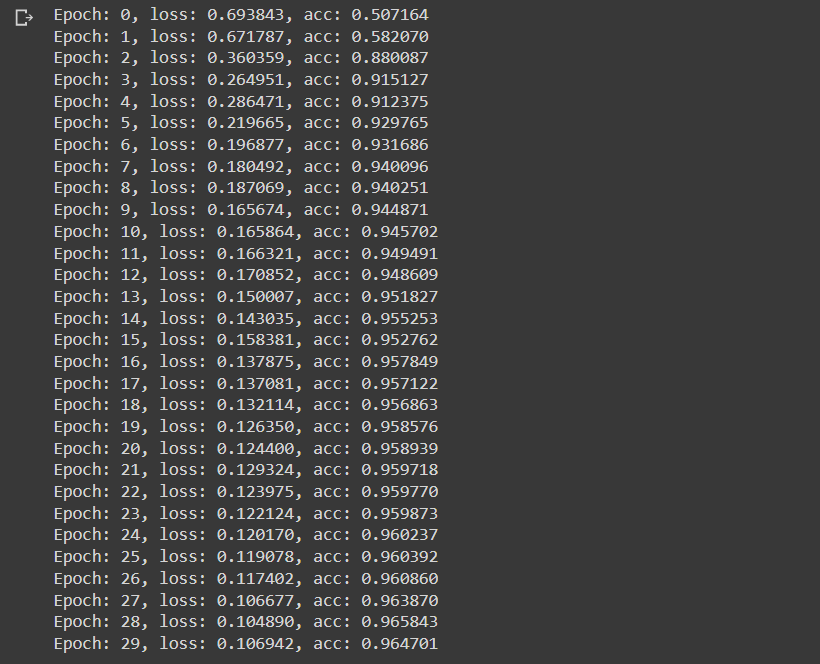
(여러 번 트레이닝을 해보니 어떨 때엔 위에서처럼 몇 epoch만에 accuracy가 확확 올라가는데 비해, 어떤 때엔 한참동안 accuracy가 오르지 않아서 트레이닝을 중단시킨적도 있는데, 뭐 때문에 이런 차이가 일어나는지는 잘 모르겠네요..)
테스트셋으로 모델의 성능을 평가해보면
# Test the model
test_loss, test_acc = 0, 0
model.eval() # state that testing is beginning (so gradients are not updated)
with torch.no_grad():
for (images, labels) in test_loader:
images, labels = images.to(device), labels.to(device)
predictions = model(images)
loss = error(predictions, labels)
test_loss += loss.to('cpu').item()
test_acc += (predictions.argmax(1) == labels).type(torch.float).to('cpu').mean().item()
test_loss /= len(test_loader) #len(test_loader) is batch size
test_acc /= len(test_loader)
print(f"Test loss: {test_loss:>6f},Test acc: {test_acc:>6f}")
95% 정확도로 적혈구에서 말라리아를 구분해낼 수 있네요! (Umer et al의 논문에 나온 모델은 데이터의 preprocessing을 거쳐서 99.98% accuracy를 보입니다..!)
직접 모델이 예측하는 것을 보고싶어,서 데이터 중에서 아무 사진이나 가져와서 모델을 적용시켜보았습니다:
img_path = 'cell_images/cell_images/Uninfected/C98P59ThinF_IMG_20150917_154235_cell_128.png'
img_array = cv2.imread(img_path)
img_original = Image.fromarray(img_array)
img_resized = img_original.resize((120,120))
img_resized
사진을 보니 정상 적혈구인 것 같네요. 모델의 예측을 한 번 보겠습니다
transform = transforms.Compose([transforms.PILToTensor()])
img_tensor = transform(img_resized).float().unsqueeze(0)
print(model(img_tensor).argmax(1))
위에서 malariadata.class_to_idx를 출력한 결과가 {'Parasitized':0, 'Uninfected':1}이었으니 이 사진에 대해 모델은 정상적혈구라고 (옳게) 판단했습니다.
재미로 사진을 하나 더 보면
img_path = 'cell_images/cell_images/Parasitized/C101P62ThinF_IMG_20150918_151335_cell_65.png'
img_array = cv2.imread(img_path)
img_original = Image.fromarray(img_array)
img_resized = img_original.resize((120,120))
img_resized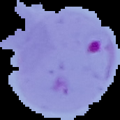
이 적혈구는 말라리아에 감염된듯 하네요.
모델의 예측을 보겠습니다:
transform = transforms.Compose([transforms.PILToTensor()])
img_tensor = transform(img_resized).float().unsqueeze(0)
print(loaded_model(img_tensor).argmax(1))
Parasitized로 (옳게) 판단했네요 (기특...).
이상 말라리아 데이터셋과 그것을 이용한 CNN모델의 소개였습니다. 컴퓨터비전에 입문하는 단계에서 MNIST 데이터셋만 가지고 공부하기 지루한 사람들에게 훌륭한 리소스인 것 같습니다!
References
1 Portrait of a serial killer | Nature
2 The top 10 causes of death (who.int)
3 IEEE Xplore Full-Text PDF:
