Needleman-Wunsch 알고리즘
-
Sequence Alignment는 한국어로 '서열 정렬'이라고 한다.
-
이에 대해 검색을 해보면 주로 '생물정보학'에서 DNA, RNA 사이의 기능적, 구조적 상관관계를 밝혀낼 때 주로 사용한다고 한다.
-
서열 정렬은 크게 두 가지로 분류할 수 있다.
-
Global Alignment : 두 시퀀스의 전체 길이에 걸쳐 최적의 정렬을 찾는 문제로 전체 구조가 유사한 시퀀스들 사이에 적용하기 적합하다.
-
Local Alignment : 두 시퀀스에서 가장 유사한 부분서열을 찾는 문제이다. 길이가 다르거나 일부 구조만 유사할 때 사용하기 적합한 방법이다.
-
-
-
Needleman-Wunsch 알고리즘은 두 시퀀스를 전역적으로 정렬하는 알고리즘으로, Global Alignment를 찾는 데 사용됩니다.
-
이 알고리즘은 동적 계획법(DP)을 기반으로 하여, 두 서열의 일치를 최적화하는 방식으로 동작합니다.
1. Needleman-Wunsch 알고리즘 개요
Global Alignment란?
-
Global Alignment는 두 서열이 전체적으로 얼마나 유사한지를 평가하는 방법입니다.
- 즉, 서열의 시작부터 끝까지 일치를 찾는 방식입니다. 이 알고리즘은 주로 두 시퀀스가 유사한 구조를 가질 때 적합합니다.
-
반대로, Local Alignment는 서열 간의 가장 유사한 부분만을 찾아내는 방식으로, Smith-Waterman 알고리즘 등이 이를 해결합니다.
동적 계획법(DP) 기반
-
Needleman-Wunsch 알고리즘은 동적 계획법(DP)을 사용하여 점수 행렬을 생성하고, 그 행렬을 채워가며 최적의 정렬을 구합니다.
-
각 셀은 세 가지 가능한 방향(대각선, 왼쪽, 위쪽) 중에서 최대값을 선택하여 채웁니다.
-
이때 각 방향의 선택은 match, mismatch, gap에 대한 점수 차이에 따라 달라집니다.
-
-
이 알고리즘의 핵심은 다음과 같다.
-
단 하나의 문제는 단 한번만 풀도록하는 알고리즘
-
한번 해결한 문제는 계속해서 다시 풀지 않고, 그 값을 저장해서 계속 사용하는 방법.
-
즉, 어떤 문제를 풀 때, 반복되는 값을 사용하는 연산이 있다면, 그 값들을 연산 돌릴때마다 새로 구하지 말고, 한번 구하면 저장해놨다가 다시 사용할 때가 되면 꺼내 쓰는 방법이라는 거다.
-
그래서, 이미 구한 값을 저장해둔다는 의미의 메모이제이션(Memoization)의 개념이 정말 중요하다.
-
-
이 알고리즘은 다음과 같은 상황에서 쓰기에 적합하다.
-
동일한 작은 문제들이 반복해서 나타나는 경우
-
부분 문제의 최적 결과 값을 사용해 전체 문제의 최적 결과를 낼 수 있는 경우
-
2. 점수 시스템 및 중요성
-
점수 설정은 알고리즘의 성능에 큰 영향을 미칩니다.
- 예를 들어, Gap penalty는 두 서열 간 공백을 삽입할 때 드는 비용을 결정하는데, 이를 조정하면 서열의 일치성을 더 잘 반영할 수 있습니다.
점수 설정
-
Match (일치): 두 문자가 같으면
+1 -
Mismatch (불일치): 두 문자가 다르면
-1 -
Gap (공백): 한 서열에서 공백을 삽입하면
-2
Gap Penalty
-
Gap penalty는 공백을 삽입할 때의 비용을 나타냅니다.
-
공백을 많이 삽입하면 점수가 많이 떨어지므로 알고리즘은 공백을 최소화하려 합니다.
-
이 값은 문제의 특성에 맞게 조정할 수 있습니다.
-
-
공백(-, 즉 gap)을 어디에 삽입하느냐에 따라
정렬의 점수가 달라지는 걸 보여주는 예시-
Case 1: X의 맨 끝에 공백을 둔 경우
X: A B C D E F G - Y: A B X D E F G W-
Match: 5개 (+1 × 5 = +5)
-
Mismatch: 6개 (-1 × 6 = -6)
-
Gap: 1개 (-2 × 1 = -2)
Alignment Score = -3
-
-
Case 2: X의 중간(_와 W 사이)에 공백을 넣은 경우
X: A B C D - E F G W Y: A B C D E F G W-
Match: 10개 (+10)
-
Mismatch: 1개 (-1)
-
Gap: 1개 (-2)
Alignment Score = +7
-
-
Match과 Mismatch
-
Match는 두 문자가 같을 때, Mismatch는 다를 때 발생합니다.
-
두 서열 간의 유사도를 평가하는 중요한 요소로, match와 mismatch 점수 설정에 따라 최종 정렬 결과가 달라질 수 있습니다.
-
예시:
Hello World&HelloWorld- 이 둘은 사람이 보기엔 “거의 같은 단어”죠. 하지만 컴퓨터 입장에서는 문자 하나라도 다르면 완전히 다른 문자열이에요.
- 그래서 이 두 문자열의 유사도(similarity) 를 계산하기 위해 정렬(Alignment) 을 시도합니다.
- 이 둘은 사람이 보기엔 “거의 같은 단어”죠. 하지만 컴퓨터 입장에서는 문자 하나라도 다르면 완전히 다른 문자열이에요.
-
핵심 정리
-
두 문자열을 어떻게 “맞춰서 정렬”하느냐에 따라 점수가 달라진다.
-
공백(gap) 의 위치는 매우 중요하다.
-
가장 높은 alignment score 를 만드는 공백 위치가 “최적 정렬(best alignment)”이다.
-
Needleman–Wunsch 알고리즘은 바로 이 최적 정렬을 자동으로 찾아주는 알고리즘이다.
-
서열 정렬 알고리즘은 “맞춤법이 맞는지”를 찾는 게 아니라, “두 문자열(또는 서열)을 가장 잘 맞춰서 비교했을 때의 ‘유사도 점수’(alignment score)”를 계산합니다.
- 즉, 점수가 높다 → 두 문자열이 매우 유사하다.
- 사람이 보기엔 “비슷하다”를 컴퓨터가 보기엔 “점수가 높다” 로 수치화한 거예요.
3. 알고리즘 실행 과정
1) 초기화
-
X와 Y 서열에 대해 점수 행렬을 초기화합니다.
-
첫 번째 행과 첫 번째 열은 gap penalty로 채웁니다.
X(i)\Y(j) 0 G C A T G C G 0 0 -1 -2 -3 -4 -5 -6 -7 G -1 1 0 -1 -2 -3 -4 -5 A -2 0 0 1 0 -1 -2 -3 T -3 -1 -1 0 1 0 -1 -2 T -4 -2 -2 -1 0 1 0 -1 A -5 -3 -3 -2 -1 0 1 0 C -6 -4 -4 -3 -2 -1 0 1 A -7 -5 -5 -4 -3 -2 -1 0
-
2) 점수 계산
-
각 셀을 채우는 과정에서
match,mismatch,gap에 대해 계산합니다.- 예를 들어,
c[1][1]은G와G를 비교하여 match 점수인1이 됩니다.
- 예를 들어,
-
Score Matrix를 만들어 Match, Mismatch, Gap이 있는 상황 중 가장 값이 높은 것으로 채워갈 것이다.
- Score Matrix는 c로 표기하겠다.
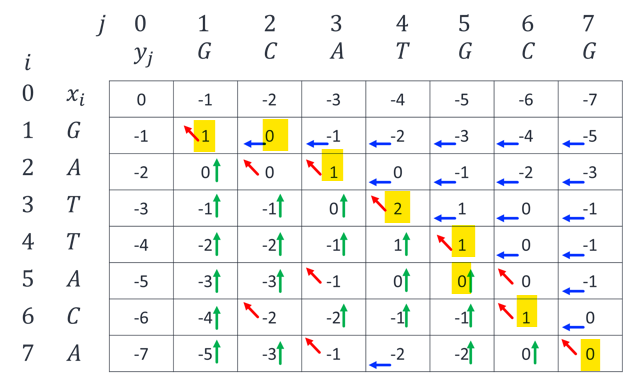
3) Backtracking
-
행렬을 채운 후, 가장 오른쪽 하단에서 시작하여 최적의 경로를 추적합니다.
- 이를 통해 서열을 정렬합니다. 대각선, 왼쪽, 위쪽으로의 이동을 추적하면서 최적의 서열을 복원합니다.
4. 알고리즘 구현 (Python)
def needleman_wunsch(X, Y, match=1, mismatch=-1, gap=-2):
m = len(X)
n = len(Y)
# 초기화: DP 테이블 생성
dp = [[0] * (n + 1) for _ in range(m + 1)]
# 첫 행과 첫 열 초기화 (gap penalty)
for i in range(m + 1):
dp[i][0] = i * gap
for j in range(n + 1):
dp[0][j] = j * gap
# 점수 계산
for i in range(1, m + 1):
for j in range(1, n + 1):
match_score = dp[i - 1][j - 1] + (match if X[i - 1] == Y[j - 1] else mismatch)
delete_score = dp[i - 1][j] + gap
insert_score = dp[i][j - 1] + gap
dp[i][j] = max(match_score, delete_score, insert_score)
# Backtracking
align_X = []
align_Y = []
i, j = m, n
while i > 0 and j > 0:
current_score = dp[i][j]
diagonal_score = dp[i - 1][j - 1]
up_score = dp[i - 1][j]
left_score = dp[i][j - 1]
if current_score == diagonal_score + (match if X[i - 1] == Y[j - 1] else mismatch):
align_X.append(X[i - 1])
align_Y.append(Y[j - 1])
i -= 1
j -= 1
elif current_score == up_score + gap:
align_X.append(X[i - 1])
align_Y.append('-')
i -= 1
else:
align_X.append('-')
align_Y.append(Y[j - 1])
j -= 1
# 남은 부분 처리
while i > 0:
align_X.append(X[i - 1])
align_Y.append('-')
i -= 1
while j > 0:
align_X.append('-')
align_Y.append(Y[j - 1])
j -= 1
# 최종 결과
return ''.join(align_X[::-1]), ''.join(align_Y[::-1]), dp[m][n]
# 테스트
X = "GCATGCG"
Y = "GATTACA"
aligned_X, aligned_Y, score = needleman_wunsch(X, Y)
print(f"Aligned X: {aligned_X}")
print(f"Aligned Y: {aligned_Y}")
print(f"Alignment Score: {score}")5. 실제 응용 및 최적화
유전자 서열 분석
-
Needleman-Wunsch 알고리즘은 DNA 서열 비교나 단백질 서열 비교에서 많이 사용됩니다.
- 예를 들어, 서로 다른 종의 유전자 서열을 비교하거나 돌연변이 지점을 찾는 데 유용합니다.
단백질 서열 비교
- 단백질 서열은 Amino Acid 배열이 중요한데, 이를 비교할 때 substitution matrix(예: BLOSUM62)를 사용하여 보다 정확한 비교를 할 수 있습니다.
시간 및 공간 최적화
-
기본적인 Needleman-Wunsch 알고리즘은 O(m × n)의 시간 복잡도와 O(m × n)의 공간 복잡도를 가집니다. 이로 인해 매우 큰 서열을 다룰 때는 성능 문제가 발생할 수 있습니다.
-
이를 해결하기 위해 Hirschberg's Algorithm이나 Affine Gap Penalties와 같은 최적화 기법을 사용할 수 있습니다.
6. 결론
-
Needleman-Wunsch 알고리즘은 두 서열 간의 전역적 정렬을 찾는 데 중요한 도구입니다.
-
동적 계획법을 통해 두 서열을 비교하여 최적의 정렬을 도출하며, 생물정보학에서 서열 분석 및 비교에 널리 사용됩니다.
-
gap penalty, match, mismatch 점수의 조정을 통해 다양한 문제에 적용할 수 있으며, 최적화 기법을 통해 계산 성능을 개선할 수 있습니다.
