[Paper Review] GraphDTI: A robust deep learning predictor of drug-target interactions from multiple heterogeneous data
Paper Review
이번에 리뷰할 논문은 Drug-target interaction을 deep learning으로 접근한 논문임. DTI에 deep learning을 적용한 논문은 상당히 많지만 이 논문은 real world에서도 유효하게 쓰일 수 있는 기법이 다소 포함되어 있는 것 같아서 리뷰함.
Liu, Guannan, et al. "GraphDTI: A robust deep learning predictor of drug-target interactions from multiple heterogeneous data." Journal of Cheminformatics 13.1 (2021): 1-17.
1 INTRODUCTION
- Drug-target interaction을 예측하는 것은 신약 개발에 있어 중요한 문제.
- Drug-target 뿐만 아니라 PPI와 differentially expressed gene(DEG)가 같이 고려되어야 함.
- DTI와 관련된 dataset은 heterogeneous한 특징을 가지고 있으며 ML을 이용한 feature-based approach가 unseen data에 대해 효과적이라고 알려져있음. 그리고 최근에는 DL을 이용한 다양한 연구가 있음.
- Challenges
- Multiple heterogeneous(e.g. information on drugs and proteins, combined or seperately, or drug-perturbed gene expression profiles and PPI neworks)를 이용하면 higher sensitivity와 lower false positive rate를 가질 것
- More carefully design validation protocols (more than random-split)
- Contributions
- 다양한 biological data를 사용함
- target protein sequences
- drug chemical structures
- the structures of drug binding sites
- the information obtained from drug-perturbed gene expression profiles
- 다른 연구들과 달리 feature selection procedure가 있임
- cluster-based split을 통해 unseen data에 대한 robustness 확보
- 다양한 biological data를 사용함
2 Results and discussion
2.1 System-level data representation and integration
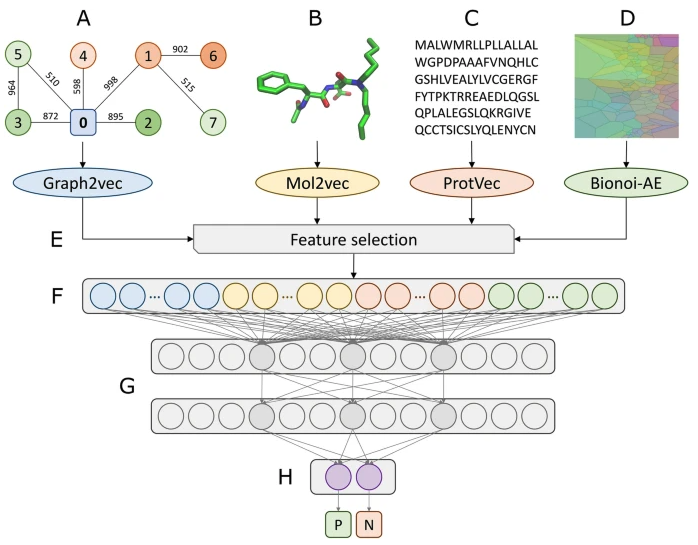
 Figure 1. Schematic representation of the local network environment for a target protein
Figure 1. Schematic representation of the local network environment for a target protein
- Combined with the PPI network information, drug-perturbed differential gene expression profiles help understand how drug binding to molecular targets alters biological processes to produce a particular phenotype
- In GraphDTI, an undirected, weighted subgraph containing a central node corresponding to the target (labeled 0 in Fig. 1) with multiple connected nodes representing interacting proteins, is extracted from the entire human PPI network
2.2 GraphDTI architecture
 Figure 2. Flowchart of GraphDTI
Figure 2. Flowchart of GraphDTI
2.3 Feature optimization for the local network environment
중요한 내용이 아니라서 pass
2.4 Feature selection with permutation feature importance
 Figure 4. Feature selection with permutation feature importance
Figure 4. Feature selection with permutation feature importance
- all 1412 features, comprising 300 drug, 300 protein, 512 drug binding site and 300 local network features, are first ranked in a descending order based on their importance scores estimated with the permutation feature importance algorithm.
- Next, the classification performance of the MLP model, pre-trained on the GraphDTI dataset, against the PubChem BioAssay dataset is calculated for a different number of the ranked features
feature가 400일때 제일 높은건.. mol2vec 중 일부가 중요한 role을 차지하거나 Bionoi-AE가 비중이 커지면서 좋은 결과를 내지 않았을까?
2.5 Visualization of the machine learning model!
 Figure 5. Separation of input features and output-layer embeddings in a low-dimensional space. A 400-dimensional input feature vectors and B output-layer embeddings prior to the softmax activate function
Figure 5. Separation of input features and output-layer embeddings in a low-dimensional space. A 400-dimensional input feature vectors and B output-layer embeddings prior to the softmax activate function
- 학습에 사용된 데이터셋이 아닌 PubChem BioAssay dataset에 대해 tSNE를 이용하여 pos/neg를 시각화함
- unseen data임에도 불구하고 pos/neg를 구분할 수 있음
2.6 Performance of DTI predictors in a random-split cross-validation
 Figure 6. Cross-validated performance of algorithms to predict DTIs.
A random-split / B cluster-based cross-validation
Figure 6. Cross-validated performance of algorithms to predict DTIs.
A random-split / B cluster-based cross-validation
- random split은 유사한 약물이나 단백질이 training / validation dataset에 모두 포함될 수 있어서 문제가 생길 수 있음
2.7 Clustering drugs and their molecular targets
 Figure 7. Optimization of the number of clusters for cross-validation
- silhouette coefficient를 통해 cluster간의 불일치도 및 cluster 내의 유사도를 측정
- scaled Perfect Match Distance를 instance간의 similarity로 정의하였을 때 silhoueete coefficinet가 가장 높게 나움
- number of clusters를 silhoueete coefficinet가 200으로 정한 후 randomly merge하여 10-fold로 만듬
Figure 7. Optimization of the number of clusters for cross-validation
- silhouette coefficient를 통해 cluster간의 불일치도 및 cluster 내의 유사도를 측정
- scaled Perfect Match Distance를 instance간의 similarity로 정의하였을 때 silhoueete coefficinet가 가장 높게 나움
- number of clusters를 silhoueete coefficinet가 200으로 정한 후 randomly merge하여 10-fold로 만듬
2.8 Performance of DTI predictors in a cluster-based cross-validation
- 기존의 방법론은 random split에 비해 cluster-based cross-validation에서 성능이 떨어지지만 GraphDTI는 계속 좋은 성능을 보여줌
- GraphDTI는 unseen data에 대해서도 robust한 성능을 보여줌
2.9 Performance of DTI predictors against unseen data
 Figure 7. Optimization of the number of clusters for cross-validation
Figure 7. Optimization of the number of clusters for cross-validation
- GraphDTI dataset에서 학습한 후 Pubchem BioAssay dataset에서도 좋은 성능을 보임
2.10 Literature validation
생략
3 Conclusion
GraphDTI 좋고 잘 만들었음ㅇㅇ
4 Materials and methods
4.1 Dataset
4.1.1 Drug-target interaction data
Positive DTI set
BindingDB에서 human protein과 drug-like properties를 가지면서 ChEMBL id가 있는 small molecule의 inteaction 선별
Negative DTI set
1) ChEMBL에서 Positive DTI set의 drug 중 Positive Tanimoto coefficient가 0.4 미만인 compound를 선별하고 2) protein sequence가 40%미만으로 겹치는 protein을 선별해서 negative DTI set을 구성함
음..? 똑같이 따라하라고 하면.. 재현하기에는 설명이 부실한듯
4.1.2 Protein-Protein interation network
- STRING database 사용
4.1.3 Differential gene expression
- CMap에서 BindingDB와 STRING database에 매칭되는 cell line-small molecule pair를 사용
- DTI set에 비해 pair의 수가 적어서 biological knowledge를 이용하여 augmentation
4.1.4 Knowledge-based data augmentation
- At high concentrations, the transcriptomic profiles of chemically similar drugs with a TC of ≥ 0.85 tend to be similar as well
- we assigned gene expression profiles from the most similar molecules with a TC of ≥ 0.85 and at the highest tested concentration
- drug similarity is measured by molecular fingerprints
- Final GraphDTI dataset
- Instance level
- Positive DTI: 326,139 instances (3618 drugs, 421 proteins, 7590 signature identifiers)
- Negative DTI: 326,188 instances (236 drugs, 358 proteins, 1541 signature identifiers)
- Unique drug-target pair level
- Positive DTI: 10,977 pair
- Negative DTI: 79,376 pair
- Instance level
4.1.5 Unseen data for independent testing
- CMap 데이터 중 BindingDB에는 없는 drug을 기준으로 PubChem BioAssay database에서 drug-target pair (195 drugs, 2152 proteins)를 추출한 후 필터링하여 unseen dataset이라 볼 수 있는 새로운 데이터셋을 구성함
4.2 Features
4.2.1 Graph-based features for machine learning
- 두 노드 사이의 shortest path상의 sum of weights of edge를 distance로 정의. top-k(fixed number of k)의 노드로 subgraph를 구성
- 구성된 subgraph를 이용하여 graph2vec으로 300dim target protein feature vector를 만들 수 있음
4.2.2 Molecular features for machine learning
- Mol2Vec을 이용한 drug feature
- ProtVec을 이용한 protein feature
- target protein의 drug binding site를 encoding한 Bionoi-AE <- 다른 DTI모델과 가지는 차별점 중 하나
- eFindSite를 이용해서 target protein의 binding site를 찾은 후 Bionoi-AE를 이용해 512dim feature vector를 만듬
4.3 Architecture and training
4.3.1 Multilayer perceptron architecture
- 다소 뻔한 얘기라서 생략
4.3.2 Feature selection
- we first assessed the accuracy score of the MLP model with original, 1412-dimensional feature vectors, denoted as 𝑆𝑜𝑟𝑖. Next, we randomly shuffled a single feature 𝑗 across all instances, without changing any other features or labels, to calculate a permutated accuracy score, 𝑆𝑝𝑒𝑟𝑚𝑗. The importance of feature 𝑗, 𝐼𝑗, is defined as
- 사실 처음 들어보는 방식. 일반적인 method는 아니고 봤더 ㄴ논문들 중 이런걸 신경쓰는건 없었음
4.3.3 Random-split cross-validation
- 다소 뻔한 얘기라서 생략
4.3.4 Clutster-based cross-validation
- k-means 대신 k-medoids를 이용하여 clustering
- 위에서도 설명했지만 scaledPMD를 distance로 사용
- he PMD [50] based on the TC [78] between drugs and the Template Modeling score (TM-score) [101] between proteins, ranging from 0 to 2‾√. Mapping all 90,353 drug-target pairs in the GraphDTI dataset to a coordinate system in the Euclidean space with the PMD puts them in a circle with a radius of 2‾√. Since this representation makes it difficult for common clustering algorithms, such as k-medoids and k-means, to work satisfactorily, we developed the following scaled version of the PMD
4.3.5 Other approaches to DTI prediction
블라블라
5 Availability of data and materials
- Code: https://github.com/Guannan1900/GraphDTI
- Dataset: https://osf.io/ugvd9/
